22 July 2020: Clinical Research
Phospholipase Cɛ Regulates Prostate Cancer Lipid Metabolism and Proliferation by Targeting AMP-Activated Protein Kinase (AMPK)/Sterol Regulatory Element-Binding Protein 1 (SREBP-1) Signaling Pathway
Yongbo Zheng1ABCDEF, Jiajia Jin2ABCDF, Yingying Gao3ACF, Chunli Luo4AG, Xiaohou Wu1AG, Jiayu Liu1ABCDE*DOI: 10.12659/MSM.924328
Med Sci Monit 2020; 26:e924328






Abstract
BACKGROUND: Metabolic reprogramming is a common characteristic of numerous kinds of tumors, including prostate cancer (PCa). Tumor metabolism such as lipid metabolism provides sufficient lipids for tumor cell division and rapid growing as well as a vital source for formation of new cellular membranes. Phospholipase Cε (PLCε) is an oncogene that can drive proliferation, progression, and lipid metabolism of tumors, but its effect in lipid metabolism of PCa is not clear.
MATERIAL AND METHODS: Benign prostatic hyperplasia (BPH) and PCa tissue specimens were assessed for SREBP-1, FASN, and PLCε by immunohistochemistry, and PLCε was knocked-down by a lentiviral short hairpin RNA. The mRNA and protein level expression of related factors were tested by qPCR and Western blot analyses. Cell proliferation was assessed by clone formation, CCK-8, and Ki-67 assays. Nile red and oil red O staining were performed to detect endogenous lipid levels. Immunofluorescence was used to localize the protein of SREBP-1. Finally, a tumor xenograft assay of nude mice was performed to assess the role of PLCε in prostate tumor generation.
RESULTS: We found that overexpression of PLCε indicates low PFS in PCa and is involved in metastasis of PCa, and that the PLCε/AMPK/SREBP-1 signaling network promotes the progression of PCa through lipid metabolism in vivo and in vitro.
CONCLUSIONS: This study is the first to discover the lethal role of PLCε in lipid metabolism and malignant behavior of PCa, elucidation PCa occurrence and progression.
Keywords: Lipid Metabolism, Phosphoinositide Phospholipase C, Prostatic Neoplasms, AMP-Activated Protein Kinases, Lipids, Prostate, RNA, Messenger, RNA, Small Interfering, Sterol Regulatory Element Binding Protein 1
Background
Prostate cancer (PCa) is the second most lethal malignancy in males worldwide [1–3], and it is also the second leading cause of cancer deaths, largely due to metastasis [4,5]. Therefore, early recognition and exploring details of the molecular metabolism to develop more effective treatments for prostate cancer are extremely important. There are a number of aspects which are intimately linked with PCa, such as age, genetic background, family history, environmental influences, diet, and lifestyle. Among these, lipid metabolism can influence many aspects of tumor biology and progression and plays a vital part in the occurrence and progression of PCa [6,7]. Epidemiologic evidence shows a relationship between obesity and progression of PCa [8]. Therefore, it is important to explore the mechanism underlying the association between lipid metabolism and prostate cancer.
Phospholipase Cɛ (PLCɛ), a phosphoinositide-specific PLC family member that has guanine nucleotide exchange factor activity. It has also been confirmed as an multifunctional signaling protein that is increased in different types of cancer [9–11] and plays a vital part in migration, proliferation, and carcinogenesis of tumors [9,12]. Of note is that our previous research has confirmed that PLCɛ was connected with lipid metabolism in castration-resistant prostate cancer (CRPC) [13], but its potential function in PCa is unclear. Sterol regulatory element-binding protein 1 (SREBP-1) is a crucial transcription factor that targets lipid metabolism-related factors such as fatty acid synthase (FASN), acetyl-CoA carboxylase (ACACA), ATP citrate lyase (ACLY), and stearyl coenzyme A desaturase 1 (SCD1) to regulate lipid metabolism, and is highly activated in cancers [14–16], including prostate cancer [17]. Whether there is a correlation between PLCɛ and SREBP-1 in fatty acid synthesis of prostate cancer is unknown. Moreover, AMPK, a gene that plays an important role in many types of cancers [18–20], was found to be closely associated with SREBP-1 in lipid metabolism. As an upstream kinase, AMPK suppresses cleavage, nuclear translocation, and transcriptional activity of SREBP-1 and SREBP-2 by directly binding and phosphorylating them, and ultimately inhibits lipogenesis and lipid accumulation in hepatocytes exposed to high levels of glucose [21]. A previous study found that the activation of AMPK by metformin or an adenosine analogue in the liver or in cultured hepatocytes can inhibit SREBP-1 expression. In the hepatic tissues of metformin-treated rats, the mRNA and protein expressions of SREBP-1 are decreased [22]. Furthermore, AMPK inhibits the synthesis of key lipogenic enzymes such as FASN and ACLY in numerous tissues by suppressing the formation of SREBP1c [23]. In fact, when activated, AMPK acts in a tumor-suppressor-like fashion [24]. Therefore, activated AMPK can simultaneously switch off 2 carcinogenic pathways (lipogenic and PI3K/mTOR pathways) [25]. However, a few studies reported the role of AMPK combined with SREBP-1 in cancers. Hence, whether the AMPK is linked with SREBP-1 in prostate cancer is unclear.
The present study explored the function of lipid metabolism in PCa through cellular and animal models. In addition, to the best of our knowledge, our study is the first to show that PLCɛ knockdown can suppress lipid metabolism and malignant behavior of PCa via the AMPK/SREBP-1 signaling pathway. Our results show that silencing PLCɛ can suppress lipid metabolism and malignant behavior in PCa through the AMPK/SREBP signaling pathway.
Material and Methods
CLINICAL PATIENTS AND TISSUE SAMPLES:
The clinical tissue samples of 60 prostate cancer (PCa) patients and 60 benign prostatic hyperplasia (BPH) patients were acquired from the First Affiliated Hospital of Chongqing Medical University (Chongqing, China) from January 2015 to July 2018. All tissues samples were confirmed to be BPH or PCa by histological examination. We collected retrospective data on patient age, tumor histological stage, Gleason score, metastasis, and serum prostate-specific antigen (PSA). The complete clinical was recorded, and clinical patient specimens used for this research were obtained with informed consent. The study was authorized by the Ethics Committee of Chongqing Medical University (Chongqing, China).
IMMUNOHISTOCHEMISTRY (IHC):
The collected tissue samples were immersed in 10% paraformaldehyde overnight and then sliced into 5-μm-thick sections. Immunohistochemical staining of BPH and PCa tissues was analyzed using the method we previously reported [13]. The sections were incubated with anti-Ki-67(Cell Signaling Technology), anti-PLCɛ (Santa Cruz), anti-p-AMPKα (Santa Cruz), anti-SREBP-1 (Abcam), or anti-FASN (Cell Signaling Technology). The data were analyzed using methods we previously reported [13].
CELLS CULTURE AND TRANSFECTION:
All human prostate cells (RWPE-1, LNCaP, and PC3) were acquired from the Chinese Academy of Sciences or American Type Culture Collection (ATCC). The PCa cell lines (LNCaP and PC3) were maintained in RPMI1640 medium with 10% fetal bovine serum (FBS), then placed in a cell incubator at 37°C with 5% CO2. The RWPE-1 cell line was cultured in keratinocyte growth medium including bovine pituitary extract (0.05 mg/ml) and human recombinant epidermal growth factor (5 ng/ml). Cells were transduced with 3 μl PLCɛ knockdown lentivirus (sh-PLCɛ#1, GCAGATATCTGATGCCATTGC; sh-PLCɛ#2, GCAGATATCTGATGCCATTGC; sh-PLCɛ#3, GGTTCTCTCCAGAA GCAACC) from Gene Pharma Company.
QUANTITATIVE REAL-TIME PCR (QPCR):
TRIzol regents were used to extract cell total RNA, and 1 ug RNA was reverse-transcribed into aliquots of double-stranded cDNA using the Prime Script™ RT reagent kit. The cDNA was amplified using the SYBR Green PCR Kit and then examined by qPCR. The primer sequences of PLCɛ, SREBP-1, FASN, ACC1, SCD1, ACACA, ACLY, PCNA, and Cyclin D1 were:
WESTERN BLOT (WB) ANALYSIS:
Total, cytoplasmic, and nuclear proteins of cells were extracted and assessed by Western blot analysis with antibodies using a standard protocol described previously [26,27]. We used the following antibodies: anti-PLCɛ (Santa Cruz); anti-PCNA, anti-Cyclin D1, anti-β-actin, anti-p-AMPKα, anti-total-AMPKα, ACLY, ACACA, SCD1 (Cell Signaling Technology), anti-SREBP-1, anti-H3 (Abcam), and FASN (Sigma).
IMMUNOFLUORESCENT STAINING:
The cells were maintained in a 6-well plate with sterile cover slip and washed with phosphate-buffered saline (PBS). Afterwards, cells were immersed in 4% paraformaldehyde for 30 minutes (min) and subsequently incubated with the primary antibody: anti-SREBP-1 (Abcam) and anti-Ki67 (Cell Signaling Technology). The experiments were performed as previously described [26].
CELL COUNTING KIT-8 (CCK-8) ASSAY:
As our previous research described [26], PCa cells receiving different treatments were maintained in 96-well plates. Optical density was examined by a microplate reader at the absorbance of 450 nm.
COLONY FORMATION TEST:
PCa cells were incubated in 6-well plates for 2 weeks and then immersed in 4% paraformaldehyde for 20 min, then stained with crystal violet for 10 min. Every treatment of cells was repeated in 3 wells and colony formation assays were repeated 3 times. CCK-8 reagent was put into each well.
OIL RED O (ORO) STAINING:
Each of cell lines were added into a 6-well plate at about 5×104 cells/well. After treatments, cells were immersed in 4% paraformaldehyde for 20 min and then washed twice with PBS. After completely drying, cells were stained with ORO solution (double-distilled water was used to dilute the original ORO solution and the ratio of water to ORO was 3: 1) for 15 min and then washed twice with PBS and photographed through a microscope. A spectrophotometer was used to quantify the lipid content at 520 nm.
NILE RED STAINING:
Nile red stain (diluted with PBS in the ratio of 1: 1000) was added to wells of a 6-well plate for lipid staining for about 20 min, then cells were washed 3 times in PBS for 5 min each time. When the 6-well plate was completely dried, DAPI was added to cells, followed by incubating for 10 min and washing twice with PBS. Image were captured using a Nikon confocal microscope.
ANIMAL STUDIES:
The experiments on animals were authorized by the Ethics Committee of Chongqing Medical University. PC3 cells were transfected with sh-NC lentivirus and sh-PLCɛ lentivirus and then injected into the right flank subcutaneous tissue of nude mice age 6 weeks. The volume of tumors was measured every week. The tumor tissue samples were removed, measured, and stored for histology studies.
STATISTICAL ANALYSIS:
All experiments were independently repeated at least 3 times. Data are presented as mean ±standard deviation (SD). Data were analyzed by SPSS version 21.0 software and GraphPad Prism version 5.0 software. Results of experiments were evaluated and analyzed using Kaplan-Meier method, one-way analysis of variance (ANOVA), two-way ANOVA, Spearman’s correlation analysis, Mann-Whitney test, and the
Results
THE PROGNOSTIC SIGNIFICANCE OF PLCɛ AND SREBP-1 EXPRESSION IN PCA:
To explore the underlying mechanism underlying the relationship between lipid metabolism and prostate cancer, tissue specimens from 60 PCa and 60 BPH were analyzed by immunohistochemistry. Immunohistochemical assays revealed that the protein expression of SREBP-1, FASN, and PLCɛ was elevated in PCa compared with BPH tissue samples (Figure 1A–1D). Meanwhile, a positive correlation was found between PLCɛ and SREBP-1 (Figure 1E) and between PLCɛ and FASN (Figure 1F) as determined by Spearman correlation analysis. The clinical characteristics and demographics of these PCa patients, as well as their relationship with the expression of PLCɛ, were generalized and summarized in Table 1. These data demonstrated that 68.3% of PCa tissue samples had a positive PLCɛ and among the various clinical parameters, histological stage (P=0.004) and bone (P=0.024) and visceral (P=0.022) metastasis were positively correlated with expression of PLCɛ. These results suggested that the excessive expression of PLCɛ is associated with PCa metastasis. In addition, Kaplan-Meier survival analysis demonstrated that the high expression of PLCɛ was associated with low progression-free survival (PFS) (95% CI, 18–27 months; median 23 months) compared with low PLCɛ (95% CI, 26–31 months; median 29 months), suggesting that high expression of PLCɛ can cause worse PFS (Figure 1G).
PLCɛ KNOCKDOWN SUPPRESSES LIPID ACCUMULATION OF PCA CELLS:
Although clinical research revealed the expression of PLCɛ was associated with SREBP-1 and FASN, it remains unclear whether PLCɛ can regulate lipid content in prostate cancer. For this purpose, we compared the fatty acid synthesis of human normal prostate epithelial cells (RWPE-1) and PCa cell lines (LNCaP and PC3). Later results showed that the lipid droplets content of RWPE-1 cells was lower than that of PCa cell lines as determined by Nile red staining (Figure 2A, 2B) and ORO staining assays (Figure 2C, 2D). Meanwhile, the protein and mRNA of PLCɛ, SREBP-1, and FASN were examined in RWEP-1 cells, LNCaP cells, and PC3 cells by Western blot and qPCR, respectively. The results revealed that the mRNA and protein levels of PLCɛ, SREBP-1, and FASN in RWEP-1 cells were significantly lower than in LNCaP and PC3 cell lines (Figure 2E, 2G). These experiments indicated that PLCɛ and the related factors of lipid metabolism were increased in PCa cells.
Subsequently, to explore the effect of upregulated PLCɛ on lipid metabolism of PCa cell lines, 3 diverse lentiviral short hairpin (Lv-sh) RNAs for PLCɛ were transfected into PCa cell lines (LNCaP and PC3), respectively, and Lv-sh-PLCɛ#3 observably decreased both protein and mRNA levels of PLCɛ as determined by Western blot and qPCR (Figure 3A–3D). The results suggested that, compared with negative control, the knocked-down PLCɛ inhibited the mRNA and protein levels of related factors of lipid metabolism and proliferation in PCa cell lines (Figure 3E–3J). Furthermore, the ORO and Nile red staining consistently revealed that knockdown of PLCɛ downregulated the level of lipid droplets (Figure 4A–4D). Ki-67, clone formation, and CCK-8 assays also demonstrated that knockdown of PLCɛ decreased the proliferation ability of PCa cells (Figure 4E–4J). Altogether, these observations suggested that knockdown of PLCɛ can inhibit both lipid metabolism and proliferation of PCa cells.
SILENCING PLCɛ BLOCKS SREBP-1 NUCLEAR TRANSLOCATION:
To further explore the underlying mechanism by which PLCɛ regulates SREBP-1 signaling in PCa, we performed follow-up experiments. Immunofluorescence assays demonstrated that silencing PLCɛ can decrease SREBP-1 expression in cell nuclei (Figure 5A). In addition, compared with negative control, the protein level of SREBP-1 was markedly downregulated in cell nuclei of prostate cancer cells after knocking down PLCɛ, as shown by Western blot (Figure 5B–5E). These results show that PLCɛ regulates lipid metabolism of prostate cancer through nuclei of SREBP-1.
:
We showed that silencing PLCɛ can suppress lipid metabolism by SREBP-1 in cell nuclei, but the underlying mechanism is unknown. Recent studies reported that some of factors can play a crucial role in lipid metabolism and irreversibly bind to SREBP-1, such as AMPK [21]. Meanwhile, a growing body of research suggests that activation of AMPK can drive prostate cancer cell death [24,28,29]. However, whether PLCɛ regulates SREBP-1 via AMPK is unknown. Thus, more detailed studies are needed to uncover the underlying mechanism. To explore this idea, a series of silencing PLCɛ were executed and then the expression of total-AMPKα and phosphorylation of AMPKα (p-AMPKα) and SREBP-1 were detected, showing that the knockdown of PLCɛ decreased the protein expression of SREBP-1 and increased p-AMPKα expression, but it did not alter the protein level of total-AMPKα (Figure 6A–6D). To further probe the effect of PLCɛ in regulating SREBP-1 by AMPK, we treated with an inhibitor of AMPK, dorsomorphin, which can suppress the phosphorylation of AMPK. Importantly, the results revealed that p-AMPK and SREBP-1 were reversed in the PLCɛ knockdown group after adding dorsomorphin (Figure 6A–6D). Furthermore, Nile red staining, clone formation, and fluorescence Ki-67 assays also showed the same results, that PLCɛ can suppress lipid metabolism and proliferation of PCa cells via AMPK/SREBP-1 (Figure 6E–6J). Taken together, these results show that PLCɛ knockdown can suppress lipid metabolism of different prostate cancer cells by AMPK/SREBP-1.
:
We found that knockdown of PLCɛ suppressed lipid metabolism and proliferation of PCa cells in vivo. We injected 2×106 of negative group or silencing PLCɛ PC3 cells (knockdown PLCɛ group) into the right flank subcutaneous tissue of each nude mice and divided them into 2 group as above. The nude mice were killed 42 days later, tumors were excised, and then assessed and measured. Compared with the negative group, the IHC results revealed that the expressions of PLCɛ, SREBP-1, and Ki-67 were reduced in the knockdown PLCɛ group but were increased in the p-AMPKα group (Figure 7A). Meanwhile, the silencing PLCɛ decreased the growth, weight, and volume of tumors compared with the control group (Figure 7B–7D). Overall, these results demonstrated that PLCɛ knockdown significantly suppressed lipid metabolism and proliferation of PCa cells in vivo.
Discussion
Finding the appropriate therapy for PCa and discovering new biomarkers to predict prostate cancer are challenging. Growing evidence indicates that the increasing number of cancer cells demands reprograming metabolism for progression and development [30], and lipogenesis is required for membrane synthesis and energy resource of cancer cells [31,32]. Previous research has reported that high levels of lipogenesis are found in different types of carcinomas, such as pancreatic cancer, breast cancer, and even prostate cancer [33–35]; therefore, suppressing the lipid metabolism may be a potential therapeutic strategy.
Our previous study indicated that PLCɛ is associated with fatty acid metabolism and has a pivotal role in proliferation of CRPC cell lines [13,26,36] but whether it affects lipid metabolism of PCa cells is poorly understood. Our previous studies also reported that PLCɛ, as an oncogene, can regulate malignant biological behavior of renal cell carcinoma and urinary bladder carcinoma of [37,38]. These studies suggested that PLCɛ is essential for the occurrence and progression of urinary system malignant tumors. Therefore, we detected the protein of PLCɛ by IHC, showing that PLCɛ level was higher and positively associated with histological stage and with bone and visceral metastasis. Meanwhile, high expression of PLCɛ indicated shorter PFS in patients with PCa. More importantly, increased PLCɛ was positively associated with fatty acid synthesis-related factors (SREBP-1 and FASN). Recent publications reported that SREBP-1 is a crucial transcriptional factor in lipid uptake and lipogenesis control [39,40] and high expression in cancers [14,15,41]. In addition, the mature SREBP-1 is translocated into the nucleus to drive fatty acid synthesis [42,43]. To explore the detailed molecular mechanism, an appropriate strategy for silencing PLCɛ of LNCap and PC3 cells was demonstrated. This study revealed that knockdown PLCɛ can suppress lipid metabolism through inhibiting SREBP-1 nuclear translocation and proliferation of LNCap and PC3 cells.
SREBP-1 is a central regulator of metabolic flux and oncogenic signaling. Recent studies have reported some genes, such as AMPK, can directly bind to or regulate SREBP-1 in lipid metabolism [21,44]. Moreover, some reports have suggested its critical role in some types of cancer [45–47], even in prostate cancer [18,48]. However, to the best of our knowledge, the function of AMPK in lipid metabolism of PCa has not been previously reported. Thus, we sought to identify whether there is a relationship among PLCɛ, AMPK, and lipid metabolism. Our study is the first to show the role of the PLCɛ/AMPK/SREBP-1 signaling pathway in the lipid metabolism of PCa. We plan to verify these results
Conclusions
This study demonstrates that elevated PLCɛ induces activation of the AMPK/SREBP-1 signaling pathway to drive prostate cancer lipid metabolism and malignant proliferation.
Figures
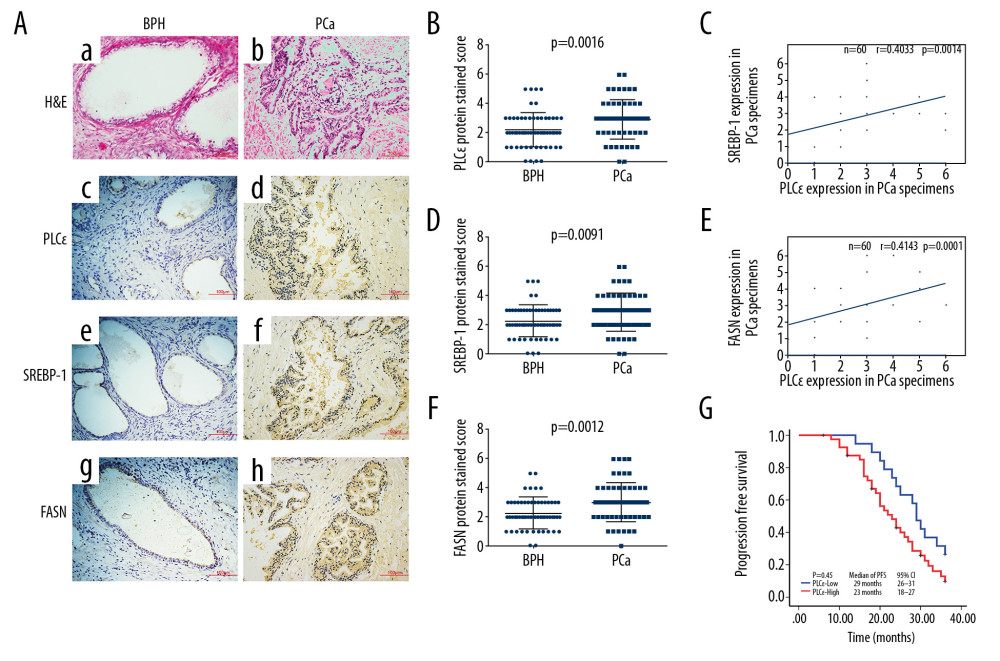 Figure 1. High expression of PLCɛ in PCa tissue specimens is related with SREBP-1/FASN. (A) Hematoxylin and eosin (HE) staining was performed on BPH and PCa tissue specimens, and PLCɛ, SREBP-1, and FASN expression levels were detected by immunohistochemistry (IHC) (200×, 100 μm bar) (a–h). (B–D) Staining scores for SREBP-1, FASN, and PLCɛ in BPH and PCa specimens. (E, F) The correlation between SREBP-1 and PLC and between FASN and PLCɛ in PCa tissue specimens was examined by Spearman analysis. (G). Progression-free survival in PCa patients was analyzed by Kaplan-Meier survival analysis.
Figure 1. High expression of PLCɛ in PCa tissue specimens is related with SREBP-1/FASN. (A) Hematoxylin and eosin (HE) staining was performed on BPH and PCa tissue specimens, and PLCɛ, SREBP-1, and FASN expression levels were detected by immunohistochemistry (IHC) (200×, 100 μm bar) (a–h). (B–D) Staining scores for SREBP-1, FASN, and PLCɛ in BPH and PCa specimens. (E, F) The correlation between SREBP-1 and PLC and between FASN and PLCɛ in PCa tissue specimens was examined by Spearman analysis. (G). Progression-free survival in PCa patients was analyzed by Kaplan-Meier survival analysis. 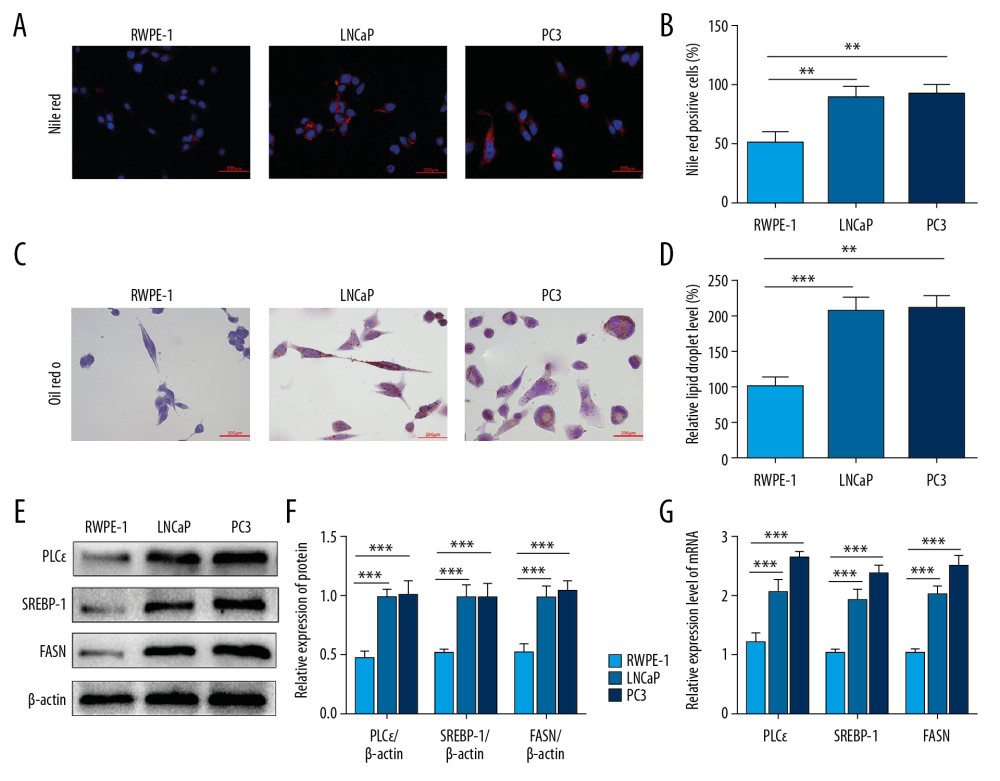 Figure 2. PLCɛ and lipid metabolism are increased in PCa cells. (A–D) Lipid droplets content level of the PCa cells (LNCaP, PC3) and RWPE-1 cells were analyzed by Nile red staining and Oil red O staining (400×, 200 μm bar). (E–G) Western blot and qPCR were performed to test the protein and mRNA expression levels of SREBP-1, FASN, and PLCɛ. The intensity of protein was quantified by image J software. Data are shown as mean±SD. *** p<0.001, ** p<0.01.
Figure 2. PLCɛ and lipid metabolism are increased in PCa cells. (A–D) Lipid droplets content level of the PCa cells (LNCaP, PC3) and RWPE-1 cells were analyzed by Nile red staining and Oil red O staining (400×, 200 μm bar). (E–G) Western blot and qPCR were performed to test the protein and mRNA expression levels of SREBP-1, FASN, and PLCɛ. The intensity of protein was quantified by image J software. Data are shown as mean±SD. *** p<0.001, ** p<0.01. 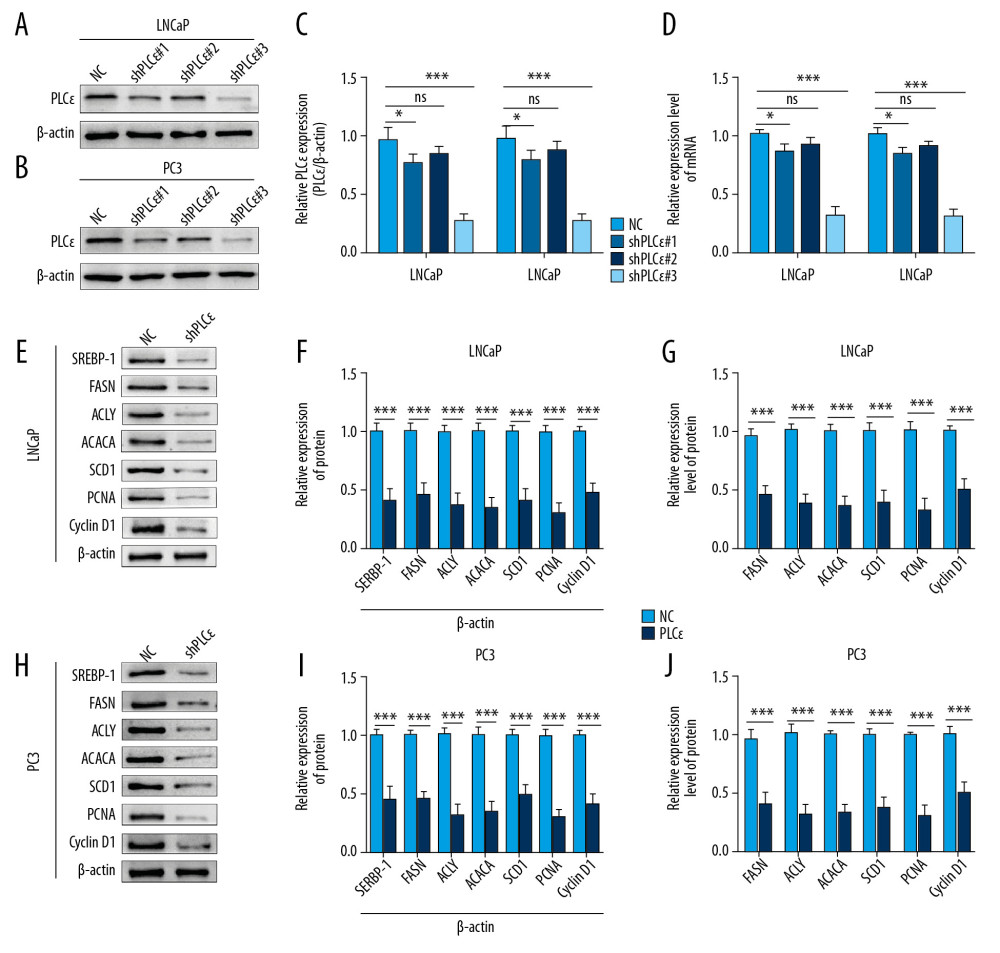 Figure 3. Silencing PLCɛ suppresses proliferation-related factors and fatty acid synthesis-related enzymes in PCa cell lines. (A–D) PC3 and LNCap cells treated with 3 types of lentivirus for knocking down PLCɛ, and the PLCɛ protein and mRNA levels were detected by Western blot and qPCR. (E–J) Knockdown of PLCɛ decreased the protein and mRNA levels of lipid metabolism-related enzymes and proliferation-related factors in PCa cells using Western blot and qPCR vs. NC.
Figure 3. Silencing PLCɛ suppresses proliferation-related factors and fatty acid synthesis-related enzymes in PCa cell lines. (A–D) PC3 and LNCap cells treated with 3 types of lentivirus for knocking down PLCɛ, and the PLCɛ protein and mRNA levels were detected by Western blot and qPCR. (E–J) Knockdown of PLCɛ decreased the protein and mRNA levels of lipid metabolism-related enzymes and proliferation-related factors in PCa cells using Western blot and qPCR vs. NC. 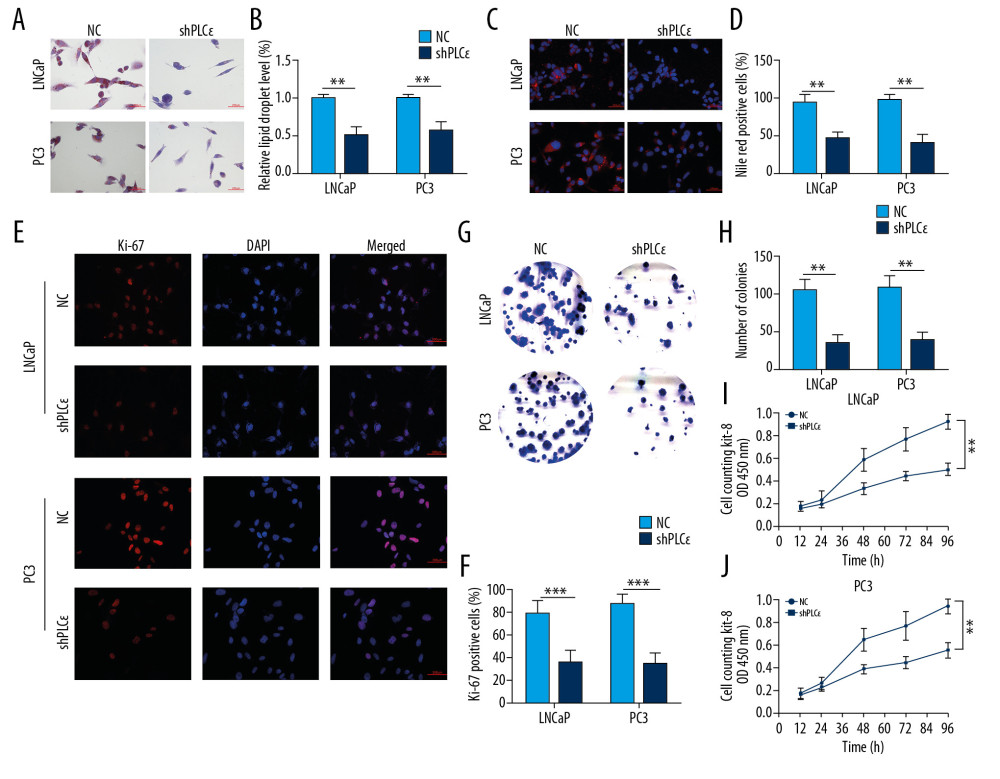 Figure 4. Silencing PLCɛ inhibits proliferation and lipid metabolism level of PCa cell lines. (A–D) The lipid droplets content of LNCaP and PC3 cells were analyzed by ORO staining and Nile red staining assays. (E–J) The fluorescence of Ki-67, clone formation, and CCK-8 assays were used to measure the proliferation of PCa cells (400×, 200 μm bar). *** p<0.001, ** p<0.01, ns – no statistical significance; NC – negative control.
Figure 4. Silencing PLCɛ inhibits proliferation and lipid metabolism level of PCa cell lines. (A–D) The lipid droplets content of LNCaP and PC3 cells were analyzed by ORO staining and Nile red staining assays. (E–J) The fluorescence of Ki-67, clone formation, and CCK-8 assays were used to measure the proliferation of PCa cells (400×, 200 μm bar). *** p<0.001, ** p<0.01, ns – no statistical significance; NC – negative control. 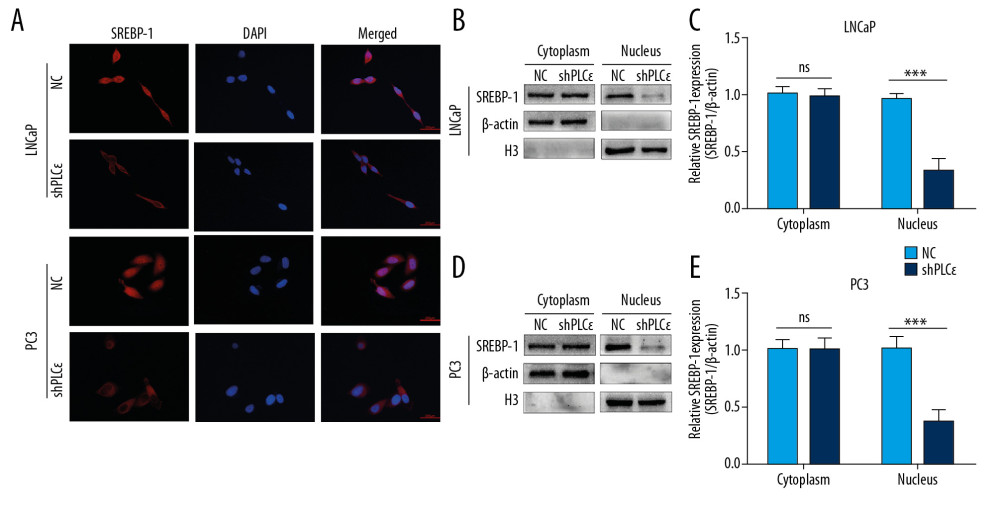 Figure 5. Silencing PLCɛ blocks SREBP-1 nuclear translocation. (A) Immunofluorescence staining revealed SREBP-1 intracellular distribution changes in LNCap and PC3 cells. (B–E) Western blot illustrated that silencing PLCɛ observably downregulates SREBP-1 expression in the nucleus but not in the cytoplasm in the 2 PCa cell lines.
Figure 5. Silencing PLCɛ blocks SREBP-1 nuclear translocation. (A) Immunofluorescence staining revealed SREBP-1 intracellular distribution changes in LNCap and PC3 cells. (B–E) Western blot illustrated that silencing PLCɛ observably downregulates SREBP-1 expression in the nucleus but not in the cytoplasm in the 2 PCa cell lines. 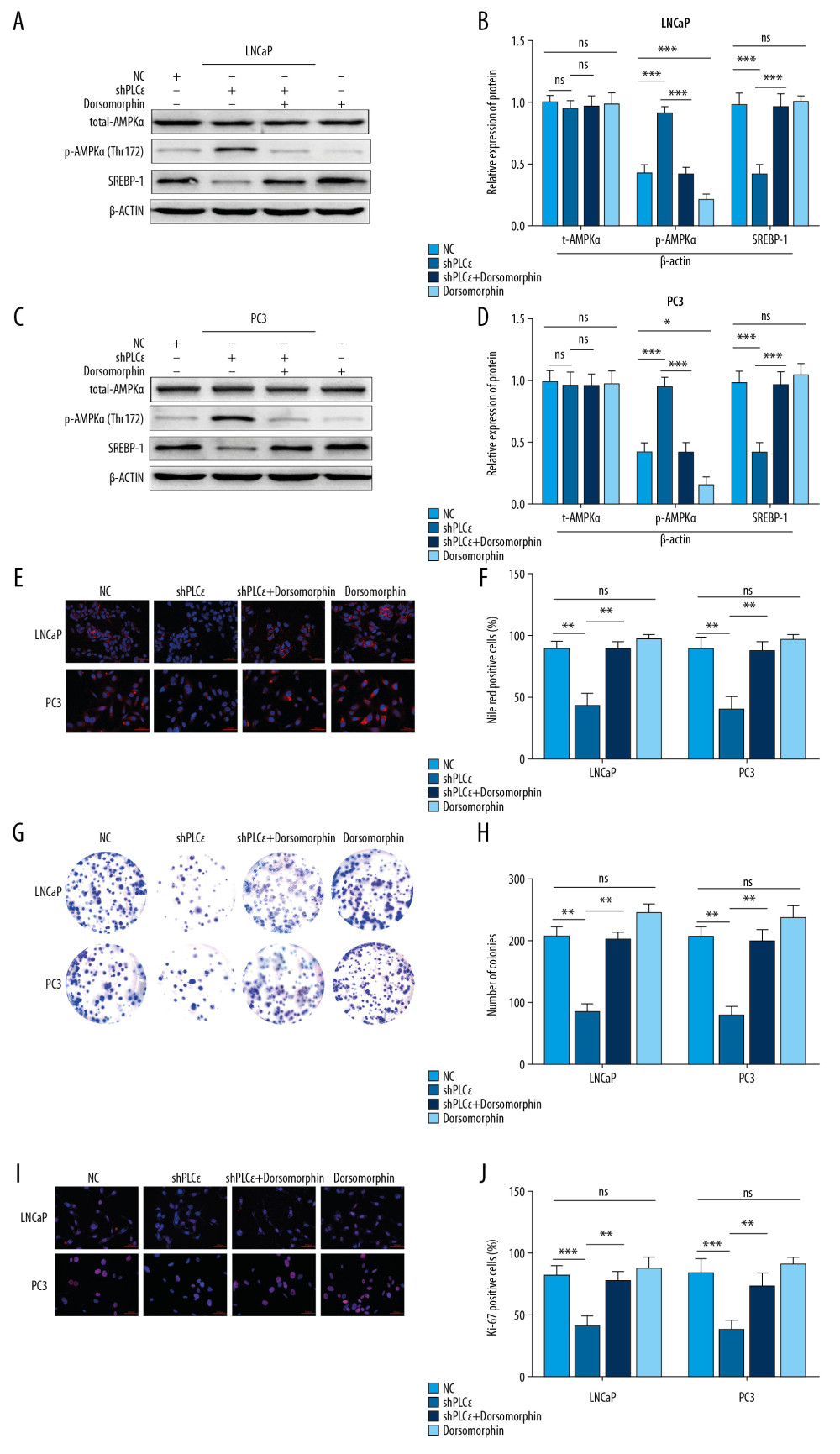 Figure 6. Silencing PLCɛ suppresses proliferation and lipid metabolism through AMPK/SREBP-1. (A–D) The protein levels of total-AMPKα, p-AMPKα, and SREBP-1 of PCa cells with different treatments, as determined by Western blot. (E, F) Nile red staining assay was used to assess lipid droplets content of PCa cells with different treatments. (G–J) Fluorescence of Ki-67 and colony formation assay were used to examine proliferation of PCa cells with different treatments. *** p<0.001, ** p<0.01, * p<0.05, ns – no statistical significance; NC – negative control. (400×, 200 μm bar).
Figure 6. Silencing PLCɛ suppresses proliferation and lipid metabolism through AMPK/SREBP-1. (A–D) The protein levels of total-AMPKα, p-AMPKα, and SREBP-1 of PCa cells with different treatments, as determined by Western blot. (E, F) Nile red staining assay was used to assess lipid droplets content of PCa cells with different treatments. (G–J) Fluorescence of Ki-67 and colony formation assay were used to examine proliferation of PCa cells with different treatments. *** p<0.001, ** p<0.01, * p<0.05, ns – no statistical significance; NC – negative control. (400×, 200 μm bar). 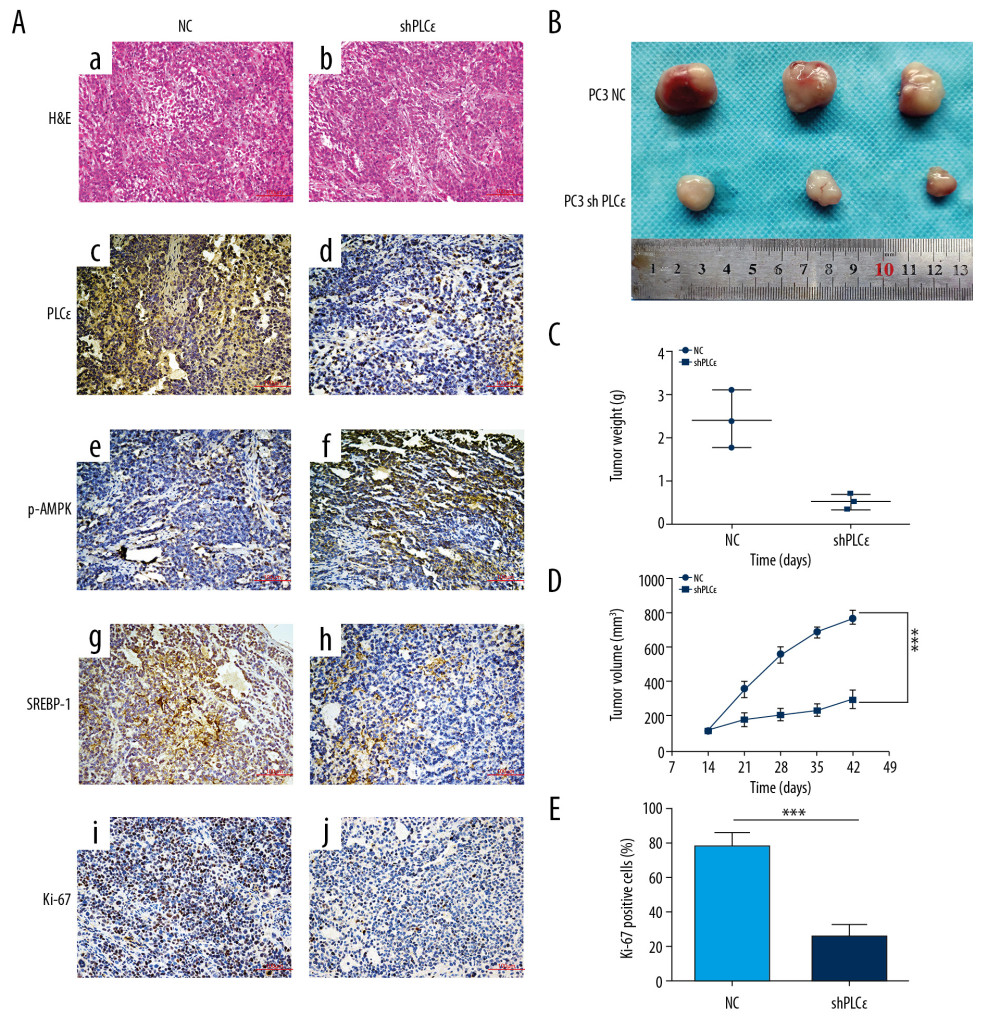 Figure 7. Silencing PLCɛ suppresses lipid metabolism and proliferation of PC3 cells in vivo. (A) Immunohistochemical assays were used to detect the expression of PLCɛ, p-AMPK, SREBP-1, and Ki67 in silenced PLCɛ and NC group (200×,100 μm bar) (a–j). (B–D) Tumor size, weight, and dynamic volume were measured. (E) Ki67 was quantified in NC and knockdown PLCɛ groups. *** p<0.001, ns – no statistical significance; NC – negative bontrol.
Figure 7. Silencing PLCɛ suppresses lipid metabolism and proliferation of PC3 cells in vivo. (A) Immunohistochemical assays were used to detect the expression of PLCɛ, p-AMPK, SREBP-1, and Ki67 in silenced PLCɛ and NC group (200×,100 μm bar) (a–j). (B–D) Tumor size, weight, and dynamic volume were measured. (E) Ki67 was quantified in NC and knockdown PLCɛ groups. *** p<0.001, ns – no statistical significance; NC – negative bontrol. 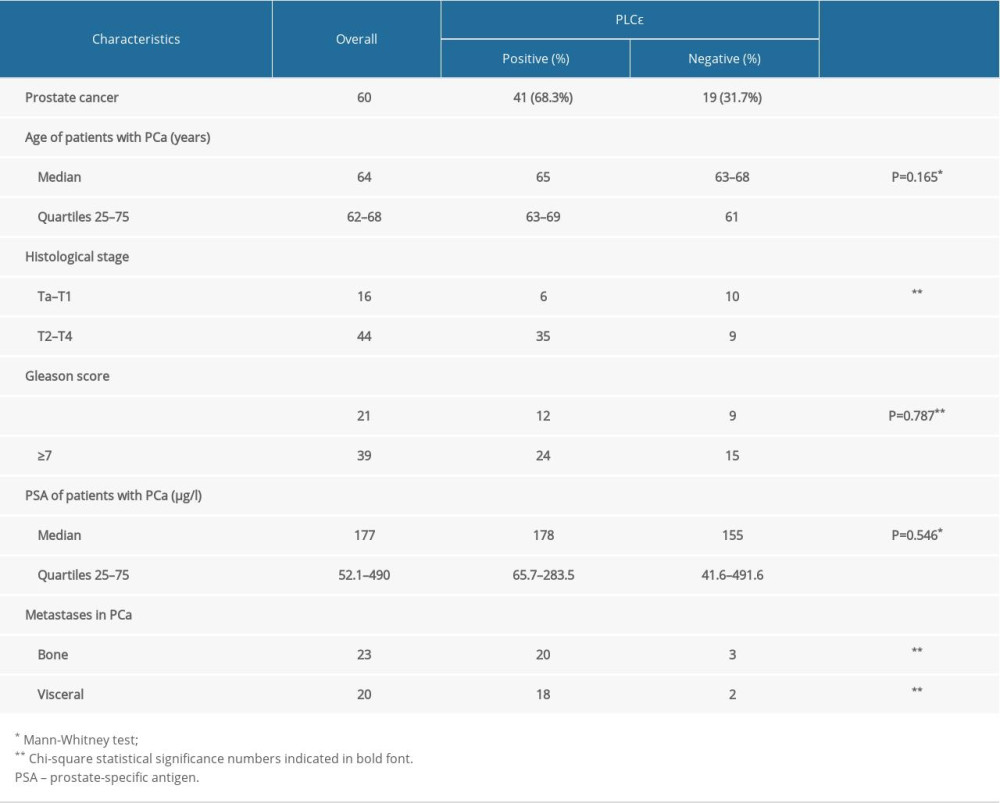
References
1. Gansler T, Ganz PA, Grant M, Sixty years of CA: A Cancer Journal for Clinicians: Cancer J Clin, 2010; 60(6); 345-50
2. Beltran H, Tomlins S, Aparicio A, Aggressive variants of castration-resistant prostate cancer: Clin Cancer Res, 2014; 20(11); 2846-50
3. Iranikhah M, Stricker S, Freeman MK, Future of bisphosphonates and denosumab for men with advanced prostate cancer: Cancer Manag Res, 2014; 6; 217-24
4. Siegel R, Naishadham D, Jemal A, Cancer statistics, 2012: Cancer J Clin, 2012; 62(1); 10-29
5. Bostwick DG, Staging prostate cancer – 1997: Current methods and limitations: Eur Urol, 1997; 32(Suppl 3); 2-14
6. Zadra G, Photopoulos C, Loda M, The fat side of prostate cancer: Biochim Biophys Acta, 2013; 1831(10); 1518-32
7. Santos CR, Schulze A, Lipid metabolism in cancer: FEBS J, 2012; 279(15); 2610-23
8. Allott EH, Masko EM, Freedland SJ, Obesity and prostate cancer: Weighing the evidence: Eur Urol, 2013; 63(5); 800-9
9. Bai Y, Edamatsu H, Maeda S, Crucial role of phospholipase Cepsilon in chemical carcinogen-induced skin tumor development: Cancer Res, 2004; 64(24); 8808-10
10. Luo XP, Phospholipase C epsilon-1 inhibits p53 expression in lung cancer: Cell Biochem Funct, 2014; 32(3); 294-98
11. Smrcka AV, Brown JH, Holz GG, Role of phospholipase Cɛ in physiological phosphoinositide signaling networks: Cell Signal, 2012; 24(6); 1333-43
12. Wang LD, Zhou FY, Li XM: Nat Genet, 2010; 42(9); 759-63
13. Gao Y, Li L, Li T, Simvastatin delays castrationresistant prostate cancer metastasis and androgen receptor antagonist resistance by regulating the expression of caveolin1: Int J Oncol, 2019; 54; 2054-68
14. Guo D, Prins RM, Dang J, EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy: Sci Signal, 2009; 2; ra82
15. Deliang G, Felicia R, Mary Y, An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway: Cancer Discov, 2011; 1(5); 442-56
16. Ettinger SL, Sobel R, Whitmore TG, Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence: Cancer Res, 2004; 64(6); 2212-21
17. Chen M, Zhang J, Sampieri K, An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer: Nat Genet, 2018; 50; 206-18
18. Lin JZ, Wang WW, Hu TT, FOXM1 contributes to docetaxel resistance in castration-resistant prostate cancer by inducing AMPK/mTOR-mediated autophagy: Cancer Lett, 2020; 469; 481-89
19. Xu Q, Wu N, Li X, Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway: Cell Death Dis, 2019; 109(12); 874
20. Guo B, Han X, Tkach D, AMPK promotes the survival of colorectal cancer stem cells: Animal Model Exp Med, 2018; 1; 134-42
21. Li Y, Xu S, Mihaylova MM, AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice: Cell Metab, 2011; 13; 376-88
22. Zhou G, Myers R, Li Y, Role of AMP-activated protein kinase in mechanism of metformin action: J Clin Invest, 2001; 108; 1167-74
23. Ferre P, Azzout-Marniche D, Foufelle F, AMP-activated protein kinase and hepatic genes involved in glucose metabolism: Biochem Soc Trans, 2003; 31; 220-23
24. Flavin R, Zadra G, Loda M, Metabolic alterations and targeted therapies in prostate cancer: J Pathol, 2011; 223; 283-94
25. Zadra G, Priolo C, Patnaik A, Loda M, New strategies in prostate cancer: Targeting lipogenic pathways and the energy sensor AMPK: Clin Cancer Res, 2010; 16; 3322-28
26. Sun W, Li L, Du Z, Combination of phospholipase Cepsilon knockdown with GANT61 sensitizes castrationresistant prostate cancer cells to enzalutamide by suppressing the androgen receptor signaling pathway: Oncol Rep, 2019; 41; 2689-702
27. Wang Y, Wu X, Ou L, PLCepsilon knockdown inhibits prostate cancer cell proliferation via suppression of Notch signalling and nuclear translocation of the androgen receptor: Cancer Lett, 2015; 362; 61-69
28. Zadra G, Photopoulos C, Tyekucheva S, A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis: EMBO Mol Med, 2014; 6; 519-38
29. Chen D, Banerjee S, Cui QC: PLoS One, 2012; 7(10); e47186
30. Ward PS, Thompson CB, Metabolic reprogramming: A cancer hallmark even warburg did not anticipate: Cancer Cell, 2012; 21; 297-308
31. Swinnen JV, Brusselmans K, Verhoeven G, Increased lipogenesis in cancer cells: New players, novel targets: Curr Opin Clin Nutr Metab Care, 2006; 9(4); 358-65
32. Mounier C, Bouraoui L, Rassart E, Lipogenesis in cancer progression (review): Int J Oncol, 2014; 45; 485-92
33. Nieva C, Marro M, Santana-Codina N, The lipid phenotype of breast cancer cells characterized by Raman microspectroscopy: Towards a stratification of malignancy: PLoS One, 2012; 7; e46456
34. Swierczynski J, Hebanowska A, Sledzinski T, Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer: World J Gastroenterol, 2014; 20; 2279-303
35. Suburu J, Chen YQ, Lipids and prostate cancer: Prostaglandins Other Lipid Mediat, 2012; 98; 1-10
36. Li L, Du Z, Gao Y, PLCepsilon knockdown overcomes drug resistance to androgen receptor antagonist in castration-resistant prostate cancer by suppressing the wnt3a/beta-catenin pathway: J Cell Physiol, 2019 [Online ahead of print]
37. Yang X, Ou L, Tang M, Knockdown of PLCepsilon inhibits inflammatory cytokine release via STAT3 phosphorylation in human bladder cancer cells: Tumour Biol, 2015; 36(12); 9723-32
38. Du HF, Ou LP, Song XD, Nuclear factor-kappaB signaling pathway is involved in phospholipase Cepsilon-regulated proliferation in human renal cell carcinoma cells: Mol Cell Biochem, 2014; 389; 265-75
39. Horton JD, Goldstein JL, Brown MS, SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver: J Clin Invest, 2002; 109; 1125-31
40. Guo D, Bell EH, Mischel P, Chakravarti A, Targeting SREBP-1-driven lipid metabolism to treat cancer: Curr Pharm Des, 2014; 20; 2619-26
41. Ettinger SL, Sobel R, Whitmore TG, Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence: Cancer Res, 2004; 64; 2212-21
42. Engelking LJ, Kuriyama H, Hammer RE, Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis: Jo Clin Invest, 2004; 113; 1168-75
43. Raghow R, Yellaturu C, Deng X, SREBPs: The crossroads of physiological and pathological lipid homeostasis: Trends Endocrinol Metab, 2008; 19; 65-73
44. Jung EJ, Kwon SW, Jung BH, Role of the AMPK/SREBP-1 pathway in the development of orotic acid-induced fatty liver: J Lipid Res, 2011; 52; 1617-25
45. Yang W, Li Y, Ai Y, Pyrazole-4-Carboxamide (YW2065): A therapeutic candidate for colorectal cancer via dual activities of Wnt/beta-catenin signaling inhibition and AMP-activated protein kinase (AMPK) activation: J Med Chem, 2019; 62; 11151-64
46. Yang L, He Z, Yao J, Regulation of AMPK-related glycolipid metabolism imbalances redox homeostasis and inhibits anchorage independent growth in human breast cancer cells: Redox Biol, 2018; 17; 180-91
47. Jiang S, Shi F, Lin H, Inonotus obliquus polysaccharides induces apoptosis of lung cancer cells and alters energy metabolism via the LKB1/AMPK axis: Int J Biol Macromol, 2019; 151; 1277-86
48. Su CC, Hsieh KL, Liu PL, AICAR induces apoptosis and inhibits migration and invasion in prostate cancer cells through an AMPK/mTOR-dependent pathway: Int J Mol Sci, 2019; 20(7); 1647
Figures
Figure 1. High expression of PLCɛ in PCa tissue specimens is related with SREBP-1/FASN. (A) Hematoxylin and eosin (HE) staining was performed on BPH and PCa tissue specimens, and PLCɛ, SREBP-1, and FASN expression levels were detected by immunohistochemistry (IHC) (200×, 100 μm bar) (a–h). (B–D) Staining scores for SREBP-1, FASN, and PLCɛ in BPH and PCa specimens. (E, F) The correlation between SREBP-1 and PLC and between FASN and PLCɛ in PCa tissue specimens was examined by Spearman analysis. (G). Progression-free survival in PCa patients was analyzed by Kaplan-Meier survival analysis.Figure 2. PLCɛ and lipid metabolism are increased in PCa cells. (A–D) Lipid droplets content level of the PCa cells (LNCaP, PC3) and RWPE-1 cells were analyzed by Nile red staining and Oil red O staining (400×, 200 μm bar). (E–G) Western blot and qPCR were performed to test the protein and mRNA expression levels of SREBP-1, FASN, and PLCɛ. The intensity of protein was quantified by image J software. Data are shown as mean±SD. *** p<0.001, ** p<0.01.Figure 3. Silencing PLCɛ suppresses proliferation-related factors and fatty acid synthesis-related enzymes in PCa cell lines. (A–D) PC3 and LNCap cells treated with 3 types of lentivirus for knocking down PLCɛ, and the PLCɛ protein and mRNA levels were detected by Western blot and qPCR. (E–J) Knockdown of PLCɛ decreased the protein and mRNA levels of lipid metabolism-related enzymes and proliferation-related factors in PCa cells using Western blot and qPCR vs. NC.Figure 4. Silencing PLCɛ inhibits proliferation and lipid metabolism level of PCa cell lines. (A–D) The lipid droplets content of LNCaP and PC3 cells were analyzed by ORO staining and Nile red staining assays. (E–J) The fluorescence of Ki-67, clone formation, and CCK-8 assays were used to measure the proliferation of PCa cells (400×, 200 μm bar). *** p<0.001, ** p<0.01, ns – no statistical significance; NC – negative control.Figure 5. Silencing PLCɛ blocks SREBP-1 nuclear translocation. (A) Immunofluorescence staining revealed SREBP-1 intracellular distribution changes in LNCap and PC3 cells. (B–E) Western blot illustrated that silencing PLCɛ observably downregulates SREBP-1 expression in the nucleus but not in the cytoplasm in the 2 PCa cell lines.Figure 6. Silencing PLCɛ suppresses proliferation and lipid metabolism through AMPK/SREBP-1. (A–D) The protein levels of total-AMPKα, p-AMPKα, and SREBP-1 of PCa cells with different treatments, as determined by Western blot. (E, F) Nile red staining assay was used to assess lipid droplets content of PCa cells with different treatments. (G–J) Fluorescence of Ki-67 and colony formation assay were used to examine proliferation of PCa cells with different treatments. *** p<0.001, ** p<0.01, * p<0.05, ns – no statistical significance; NC – negative control. (400×, 200 μm bar).Figure 7. Silencing PLCɛ suppresses lipid metabolism and proliferation of PC3 cells in vivo. (A) Immunohistochemical assays were used to detect the expression of PLCɛ, p-AMPK, SREBP-1, and Ki67 in silenced PLCɛ and NC group (200×,100 μm bar) (a–j). (B–D) Tumor size, weight, and dynamic volume were measured. (E) Ki67 was quantified in NC and knockdown PLCɛ groups. *** p<0.001, ns – no statistical significance; NC – negative bontrol. In Press
14 Mar 2024 : Clinical Research
Differential DHA and EPA Levels in Women with Preterm and Term Births: A Tertiary Hospital Study in IndonesiaMed Sci Monit In Press; DOI: 10.12659/MSM.943895
15 Mar 2024 : Clinical Research
Evaluation of an Optimized Workflow for the Radiofrequency Catheter Ablation of Paroxysmal Atrial FibrillationMed Sci Monit In Press; DOI: 10.12659/MSM.943526
09 May 2024 : Review article
A Review of the Current Status of Disease-Modifying Therapies and Prevention of Alzheimer’s DiseaseMed Sci Monit In Press; DOI: 10.12659/MSM.945091
09 Apr 2024 : Clinical Research
Correlation between Thalamocortical Tract and Default Mode Network with Consciousness Levels in Hypoxic-Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.943802
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952