05 July 2021: Clinical Research
Identification and Characterization of Non-Coding RNAs in Thymoma
Guanglei Ji1AE, Rongrong Ren2BCDF, Xichao Fang3BCDF*DOI: 10.12659/MSM.929727
Med Sci Monit 2021; 27:e929727






Abstract
BACKGROUND: Thymoma is the most common tumor of the anterior mediastinum, and can be caused by infrequent malignancies arising from the epithelial cells of the thymus. Unfortunately, blood-based diagnostic markers are not currently available. High-throughput sequencing technologies, such as RNA-seq with next-generation sequencing, have facilitated the detection and characterization of both coding and non-coding RNAs (ncRNAs), which play significant roles in genomic regulation, transcriptional and post-transcriptional regulation, and imprinting and epigenetic modification. The knowledge about fusion genes and ncRNAs in thymomas is scarce.
MATERIAL AND METHODS: For this study, we gathered large-scale RNA-seq data belonging to samples from 25 thymomas and 25 healthy thymus specimens and analyzed them to identify fusion genes, lncRNAs, and miRNAs.
RESULTS: We found 21 fusion genes, including KMT2A-MAML2, HADHB-REEP1, COQ3-CGA, MCM4-SNTB1, and IFT140-ACTN4, as the most frequent and significant in thymomas. We also detected 65 differentially-expressed lncRNAs in thymomas, including AFAP1-AS1, LINC00324, ADAMTS9-AS1, VLDLR-AS1, LINC00968, and NEAT1, that have been validated with the TCGA database. Moreover, we identified 1695 miRNAs from small RNA-seq data that were overexpressed in thymomas. Our network analysis of the lncRNA-mRNA-miRNA regulation axes identified a cluster of miRNAs upregulated in thymomas, that can trigger the expression of target protein-coding genes, and lead to the disruption of several biological pathways, including the PI3K-Akt signaling pathway, FoxO signaling pathway, and HIF-1 signaling pathway.
CONCLUSIONS: Our results show that overexpression of this miRNA cluster activates PI3K-Akt, FoxO, HIF-1, and Rap-1 signaling pathways, suggesting pathway inhibitors may be therapeutic candidates against thymoma.
Keywords: Sequence Analysis, RNA, Thymus Neoplasms, Biomarkers, Tumor, Thymoma
Background
The thymus, a small organ located behind the breast bone and just in front of and above the heart, is the primary site of T cell development and plays an essential role during the development of the immune system, including T cell differentiation and maturation [1]. Common signs and symptoms of tumors in the thymus include shortness of breath, cough with or without hemoptysis, chest pain, trouble swallowing, loss of appetite, and weight loss. Some thymoma diagnostic methods are chest X-rays, CT scans, MRIs, PET scans, biopsy, and the Chamberlain procedure [2]. Blood-based diagnostic markers are not currently available, but they would help to detect myasthenia gravis (MG) or other associated autoimmune disorders. The standard treatments available against thymoma and thymic carcinoma are surgery, radiation therapy, chemotherapy, hormone therapy, targeted therapy, and immunotherapy (under clinical trial) [3,4].
Thymic epithelial tumors (TETs) are infrequent malignancies arising from the epithelial cells of the thymus of middle-age patients; thymoma is the most predominant and clinically aggressive TET [5]. TETs are the most common tumors in the anterior mediastinum, but are infrequent in comparison to other thoracic tumors [6,7]. According to the American Society of Clinical Oncology (ASCO), thymomas account for 20% of all anterior mediastinal tumors, with an overall incident rate lower than 1 per 1.5 million, and a 5-year survival rate of 71% [50]. However, the survival rate differs depending on various factors such as cancer classification and stage. Thymomas are characterized by their unique association with autoimmune diseases and a thymic carcinoma [5]. According to the WHO, thymomas are histologically classified into 6 types: A, AB, B1, B2, B3, and C. Types A and AB are considered benign, with low mortality and rate of growth, whereas types B1, B2, and B3 are more aggressive and display a high propensity for spreading intrathoracically [8].
Thymomas have been associated with several paraneoplastic conditions, and a variety of immune and non-immune and endocrine disorders [9]. The most common syndromes associated with thymoma are myasthenia gravis (MG) (in ~30% of patients), pure red cell aplasia (PRCA) (in 5–10% of patients), and hypogammaglobulinemia (in 3–6% of patients). MG is characterized by development of autoimmune antibodies to the acetylcholine receptor in the postsynaptic neuromuscular junction [10], PRCA occurs due to immune-mediated suppression of erythropoiesis [11], and hypogammaglobulinemia occurs due to a collective deficiency of B and T cells that lead to an increased risk of infection [12]. In addition, thymomas are associated with other paraneoplastic conditions like polymyositis, systemic lupus erythematosus, limbic encephalitis, sensory-motor neuropathy, rheumatoid arthritis, thyroiditis, ulcerative colitis, acute pericarditis, myocarditis, hemolytic anemia, Addison’s disease, and Cushing’s syndrome [10]. The thymus plays an important role during development and is very sensitive to both acute and chronic injury. The thymus goes through rapid degeneration as a result of toxic insults and due to aging at a faster rate than other tissues. However, it is capable of regenerating and restoring its function to a certain degree by a mechanism involving keratinocyte growth factor (KGF) signaling [1].
Recent advances in high-throughput technologies, such as next-generation sequencing (NGS), have revealed that a significant portion of the genome (~75%) is transcribed to non-coding RNAs (ncRNAs). Among the ncRNAs, the long ncRNAs (lncRNAs) are usually at least 200 nucleotides long, while small RNAs (such as miRNAs and siRNAs) are <200 nucleotides long. According to the ENCODE (ENCyclopedia of DNA Elements) Project Consortium (GENCODE release 34), the human genome contains around 19 959 protein-coding genes, 17 960 lncRNAs genes, and 7578 small ncRNAs [13]. lncRNAs are generally mRNA-like transcripts that have conserved secondary structures and lack long open reading frames (ORFs) and have low sequence conservation. Hence, they are difficult to detect computationally from genome sequences [14]. The roles of lncRNAs in genomic regulation are multiple, involving transcriptional and post-transcriptional regulation, and imprinting and epigenetic modifications. Studies have documented that lncRNAs play key roles during tumorigenesis and tumor progression [15–17]; in particular, aberrant lncRNA expression or lncRNA dysfunction may lead to DNA damage, immune escape, and cellular metabolic disorders in cancer cells. lncRNAs can regulate cell proliferation, apoptosis, migration, invasion, and maintenance of stemness during the development of cancer. Moreover, many lncRNAs are transcriptionally regulated by key tumor suppressors or oncogenes [15]. The inherent heterogeneity and diversity of the lncRNA world make the complex tumorigenesis process more interesting than it would otherwise be [17]. Hence, identification and characterization of lncRNAs could open the doors to new clinical approaches to understand complex tumorigenesis and treat cancers.
Gene fusions occur due to recombination of 2 unrelated genes through chromosomal rearrangements or translocations. Gene fusions are key drivers of human cancer development, and these appear to play significant roles in the aggressive behavior of cancers [18].
In this study, we attempted to identify and characterize fusion genes, lncRNAs, and miRNAs from RNA-seq data of patients with thymoma, identifying aberrant lncRNAs and miRNAs in thymoma, constructing a lncRNA-mRNA-miRNA regulatory network in thymoma, and performing a functional enrichment analysis.
Material and Methods
DATA COLLECTION AND READ QUALITY CHECK:
We used 25 samples of Illumina paired-end RNA-Seq data of cancerous thymomas for this study; the same is available on the NCBI-SRA website (https://www.ncbi.nlm.nih.gov/sra) with a SRA run series from SRR1296011 to SRR1296035. We also used healthy thymus RNA-Seq data available at a bearing run series, SRR4175275, SRR4175279, SRR4175280, SRR6753058 to SRR6753061, SRR6753146 to SRR6753153, and SRR6753211 to SRR6753220 (25 samples). Moreover, we downloaded small RNA-Seq datasets of patients with thymic malignancy and normal thymus tissues from the BioProject Accession PRJNA317556 website (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA317556). We collected data from 11 thymic malignancy samples belonging to different cancer stages and 9 normal thymus gland specimens (Table 1). The raw sequence reads output from the sequencer were subjected to quality checks. We checked the quality of the RNA-Seq data in FASTQ format using the FastQC software tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) [19]. The quality check results showed that all the sequence reads belonging to various sample types were of high quality (Q-score >20) and did not contain adapters.
We developed a methodological pipeline to process RNA-Seq data for the detection of fusion genes, lncRNAs, and small ncRNAs (miRNAs). Figure 1 depicts the downstream analysis along with the set of tools used. The detailed methodology is described below.
FUSION GENE DETECTION:
Fusion genes, formed by chromosomal breakage and rejoining events, are the most common mutation class in cancers. They result from chromosomal rearrangements including translocation, inversions, deletions, and duplications [20–22]. Fusion genes lead to chimeric transcripts that deregulate genes through juxtaposition of novel promoter or enhancer regions [21]. Common methods for fusion gene detection are fluorescence in situ hybridization (FISH), RT-PCR, and next-generation sequencing (NGS). The FISH and RT-PCR methods are used for a single gene fusion and cannot detect fusion partners or complex structural rearrangements [22]. Moreover, these 2 techniques are poorly sensitive to intra-chromosomal fusion gene detection [23,24]. The third technique (NGS) is now a popularly used method for fusion detection at the whole-genome and transcriptome scale; it employs RNA-Seq data from NGS [25]. RNA-seq are mostly used because they only require a sequence region (targeted region) of the genome to be transcribed and split into mature mRNAs, making it quick and cost-effective, while it can detect multiple fusion genes.
A plethora of fusion detection tools has been developed during the last decade, including deFuse [26], EricScript [27], JAFFA [28], STAR-Fusion [29], and SQUID [30] tools. These tools are conceptually classified into 2 groups [31]: i) mapping-first approaches, in which RNA-seq reads are first aligned to the reference genome to detect discordant mapping reads suggestive of rearrangements, and ii) assembly-first approaches, in which assembled reads are followed by identification of chimeric transcripts. In this study, we used STAR-Fusion release v1.8.0 available at https://github.com/STAR-Fusion/STAR-Fusion/ to investigate therapeutic implications using RNA-seq data to detect fusion genes in both thymoma and healthy thymus datasets. We selected STAR-Fusion due to its outstanding performance published in one of the benchmarking studies among 23 fusion detection tools for cancer transcriptomes [31]. STAR-Fusion belongs to the Trinity Cancer Transcriptome Analysis Toolkit (CTAT) project, which takes Illumina RNA-seq reads as input and generates a list of candidate fusion genes as output. STAR-Fusion, a mapping-first approach, uses the STAR Aligner [32] that generates all the chimeric splits and discordant reads used as the input for the STAR-Fusion. STAR-Fusion maps the reads to exons of reference gene annotations and co-ordinates overlaps (Figure 1).
READ MAPPING AND TRANSCRIPT ASSEMBLY:
The clean RNA-seq data of both 25 patients with thymoma and 25 healthy individuals were first mapped to the Homo sapiens hg19 reference genome (downloaded from UCSC Genome Browser http://hgdownload.soe.ucsc.edu/downloads.html#human) using a recently updated version 2.4.0 of the Bowtie2 [33] and Tophat 2 version 2.1.1 [34] read mappers (Figure 1). The results of the read mapping were obtained in BAM/SAM format, with 1 file for each sample. To obtain a transcript assembly, we used Cufflinks, which is a widely used tool [35]. The Cufflinks tool assembles individual transcripts aligned to the genome, estimates abundance, and quantifies the expression level of genes in the form of FPKM (fragments per kilobase of exon model per million reads mapped). Multiple RNA-Seq samples were pooled and the assembled transcripts were merged into a comprehensive set of transcripts before the downstream analysis. For this purpose, we used Cuffmerge, which is a meta-assembler [35,36] that merges all the assemblies parsimoniously.
LONG NON-CODING RNA DETECTION:
After the quality check, read mapping, and transcript assembly, we filtered all the transcripts having a length of 200 bp or more to detect lncRNAs. We used the FEELnc (FlExible Extraction of LncRNAs) [37] tool to detect and annotate lncRNAs from RNA-seq assembled transcripts (Figure 1). FEELnc is a Random Forest-based model trained using general features such as k-mer frequencies and relaxed open reading frames (ORFs). The FEELnc is an all-in-one package that allows filtering of non-lncRNA-like transcripts and computation of coding potential scores. Figure 1 depicts the complete pipeline to detect lncRNAs.
MIRNA DETECTION AND EXPRESSION PROFILING:
MicroRNAs (miRNAs) are short non-coding RNAs (~22 nucleotides in length) which inhibit gene regulation through base-pairing at 3′ or 5′-untranslated regions of mRNAs [38]. These regulate more than 30% of the genes expressed in animal genomes [39]. A single miRNA may have multiple targets, and a single mRNA can be regulated by several miRNAs. The regulatory potential of miRNAs affects nearly all physiological processes, including cell growth, cell differentiation, and apoptosis. Many human cancers have been associated with the dysregulation of certain miRNAs [40]. The expression profile of miRNAs differs significantly between cancerous and non-cancerous tissues, and between aggressive and asymptomatic cases. miRNAs have been implicated as tumor suppressors of some cancers. Moreover, miRNAs may be good diagnostic markers; ie, miRNA expression profiles can be used to classify cancers [41].
For this study, we used the miRDeep* [42] software tool to detect miRNAs and their expression profiles in thymoma and healthy thymus tissues. MiRDeep* is a powerful tool for the detection of known and novel miRNAs from small RNA-seq data. Both known and novel miRNA expression profiles portray RNA-seq reads relative to pre-miRNA hairpins. Table 1 present the small RNA-seq data we used.
LNCRNA-MIRNA-MRNA NETWORK AND FUNCTIONAL ENRICHMENT ANALYSES:
To construct a lncRNA-mRNA-miRNA regulatory network, we considered gene co-expression association among the top-ranked lncRNAs and mRNAs using Pearson’s correlation coefficient (R2 ≥0.7). We searched for genes with similar expression patterns to those of lncRNAs in thymoma tissues using GEPIA 2.0 (http://gepia2.cancer-pku.cn/), based on TCGA tumor, TCGA normal, and/or GTEx expression data for the detection of similar genes. Next, we retrieved the target genes of selected miRNAs with target-scores ≥75 from the miRDB database [43]. We used the Cytoscape v3.8 software (https://cytoscape.org/) to visualize the lncRNA-miRNA-mRNA networks constructed based on intersections of co-expressed lncRNAs, miRNAs, and mRNAs. In addition, we performed functional and pathway enrichment analyses of target genes of DELs and DEMs using the DAVID 6.8 tool (https://david.ncifcrf.gov/), which identifies gene classes that are over- or under-represented with the GO terms, and that have associations with disease phenotypes [44,45].
Results
FUSION GENE DETECTION:
Out of 25 thymoma samples, we observed 21 fusion events in 9 samples (36%, 9/25), and 1 sample had multiple types of fusion genes. The identified fusion genes in thymus cancer and normal samples are shown in Table 3. Our results show that 5 out of 21 identified fusion genes are significant and unique to thymomas, namely KMT2A-MAML2 (detected in 9 samples of thymoma and 1 sample of the healthy thymus), HADHB-REEP1 (detected in 6 samples of thymoma), COQ3-CGA (detected in 6 samples of thymoma, and 1 sample of healthy thymus), MCM4-SNTB1 (detected in 5 samples of thymoma), and IFT140-ACTN4 (detected in 4 samples of thymoma). Table 3 presents the fusion chromosomes and breakpoints. To further analyze the results, we created a Circos plot [46] of these 5 significant fusion genes (Figure 2) illustrating genomic rearrangements and the associations between 2 genomic positions. To validate the identified fusion genes computationally, we searched the junction sequence (identified by the STAR-Fusion) in the sequence reads of the samples and confirmed their presence. We used a methodology similar to that described by Singh and Li [47].
IDENTIFICATION OF ABERRANT LNCRNAS:
To detect the aberrantly expressed lncRNAs in thymoma, we performed a genome-wide lncRNA differential expression study between thymoma and healthy thymus tissues. We obtained a list of 65 differentially-expressed lncRNAs over the 2 sample types (P value <0.05 and log of fold-change (logFC) >2.0) (Table 4), out of which 6 lncRNAs had a |logFC| >5.0. To validate the significance of the differentially-expressed lncRNAs (DELs) in thymoma using an independent tertiary dataset, we used the TCGA (The Cancer Genome Atlas) database. We compared the expression levels of 6 lncRNAs (P value=0, FDR=0, logFC >5.0), namely AFAP1-AS1, LINC00324, ADAMTS9-AS1, VLDLR-AS1, LINC00968, and NEAT1, between thymoma (THYM) and normal tissues (Figure 3), and found them to be significantly dysregulated in thymomas. To further extend our analysis, we assessed the disease-free survival (DFS) of patients with high- and low-expression groups using the Mantel-Cox test. We found that both low and high expressions of these lncRNAs directly affect patient survival. Among these 6 lncRNAs, the high expression of AFAP1-AS1 and low expression of LINC00324 greatly affected the survival of thymoma patients. Figure 4 presents a DFS map showing the survival contribution of the top 25 dysregulated lncRNAs in various cancers, including thymoma (THYM), using TCGA datasets with a significance level of 0.05 estimated using the Mantel-Cox test. The DFS map in Figure 4 shows that aberrant expression of these 6 lncRNAs can be used to characterize thymomas from other cancers.
MIRNA DETECTION AND ANALYSIS:
To detect miRNAs and analyze differentially-expressed miRNAs (DEMs), we processed the datasets listed in Table 1. A significant number of miRNAs were overexpressed in the thymomas and their expressions were absent in the healthy thymus tissues. We carried out 1695 miRNA expression profiles. We detected 294 DEMs (|logFC| ≥1.0); 198 miRNAs were upregulated and 96 were downregulated in thymomas (Supplementary Table 1 lists all the DEMs we found). Among the 294 DES, 10 miRNAs had |logFC|>10.0; Figure 5 shows their boxplot and a significant difference in the expression between thymomas and normal tissues. Figure 6 shows the logFC parameters of the top 50 DEMs with most DEMs upregulated with a maximal logFC of 15, and a few downregulated DEMs with a maximal logFC of 10.
FUNCTIONAL ANNOTATION OF THE TOP 10 RANKED LNCRNAS:
We examined the functional annotation of the top 10 ranked lncRNAs using the DAVID 5.8 tool. Each lncRNA was found to be associated with specific functions. For example, LINC00324 and FAM87A were associated with the sequence feature of putative uncharacterized protein C17orf44, and its transmembrane region, respectively. Also, LINC000324 was tissue-enriched with GO terms Brain and Lymph; FAM87A was tissue-enriched with the Go term Stomach; and HSD52 was tissue-enriched with the Go term Testis. Moreover, VLDLR-AS1 and LINC00324 were associated with embryo development, thyroid tumor disease, germ cell tumor disease, and others; while NEAT1 was associated with adrenal tumor disease, thyroid tumor disease, and others.
Furthermore, we used the UCSC-TFBS algorithm of the DAVID tool to study protein interactions including transcription factors (TFs) with a set of target genes. Out of the top 10 ranked lncRNAs, 4 (AFAP1-AS1, VLDLR-AS1, LINC00324, and HSD52) are involved in protein interactions and have functions related to TFs. Table 5 summarizes the functional annotations.
DIFFERENTIALLY-EXPRESSED LNCRNA-MRNA-MIRNA INTERACTION NETWORK:
We constructed an lncRNA-mRNA-miRNA regulation network based on the gene co-expression correlation analysis between DELs, mRNAs, and DEMs identified in thymomas (Figure 7). In this network, the upregulated miRNA hsa-let-7a-3 exhibits interactions with 8 protein-coding genes (INSR, IGF1, IL10, IGF1R, ITGB3, COL5A2, ZNF322, PXDN, TGFBR1) and can increase their expressions. The majority of target genes of DELs and DEMs are enriched in several biological pathways, including the PI3K-Akt signaling pathway (P value=0.0), the FoxO signaling pathway (P value=0.0), the HIF-1 signaling pathway (P value=0.0), the proteoglycans in cancer (P value=0.01), and other cancer pathways (P value=0.01), the Supplementary Table 2 shows these details. Most target genes were associated with various GO terms, including immune response (GO: 0006955), hepatic immune response (GO: 0002384), positive regulation of DNA replication (GO: 0045740), positive regulation of cell proliferation (GO: 0008284), positive regulation of MAPK cascade (GO: 0043410), positive regulation of DNA replication (GO: 0045740), positive regulation of cell proliferation (GO: 0008284), negative regulation of extrinsic apoptotic signaling pathway (GO: 2001237), and negative regulation of apoptotic process (GO: 0043066). Supplementary Table 3 shows these details. Our network and pathway analyses show the overexpression of miRNA clusters activates the PI3K-Akt/FoxO/HIF-1/Rap-1 signaling pathway, suggesting that PI3K/Akt/HIF-1/Rap-1 inhibitors may be therapeutic targets for thymoma patients. The hsa-let-7a family of miRNAs is thought to inhibit migration, invasion, and tumor growth by targeting the AKT2 in papillary thyroid carcinoma [48]. Increased plasma levels of hsa-let-7 have been found in patients with breast, prostate, colon, renal, liver, gastric, ovarian, and thyroid cancers [49].
Discussion
Thymomas, predominant and clinically aggressive among the thymic epithelial tumors, are the most common tumors in the anterior mediastinum, but are infrequent in comparison to other thoracic tumors. The thymus is the primary site of T cell development and plays an essential role in the development of the immune system, T cell differentiation, and maturation [1]. The thymus has been shown to be associated with several paraneoplastic conditions, and a variety of immune and non-immune and endocrine disorders [9]. One-third of patients with thymus malignancy are mostly asymptomatic during their initial stages. However, as the malignancy progresses, patients start to present signs such as cough, chest-pains, nerve palsy, and superior vena cava syndrome [50]. Engels reported some major related cancers that can form due to thymoma, including lung cancer, thyroid cancer, prostate cancer, leukemia, and sarcomas [51]. Liu et al [52] indicated in their case study that an elderly patient suffering from acute gastric volvulus (AGV) had a primary thymoma, and they hypothesized that thymoma/thymus cancers can develop either individually or underlying a secondary disease.
Genomic instability (ie, gene mutations, translocations, deletions, copy number alterations, and inversions) can lead to cancer and other diseases. These genetic events can also trigger gene fusions. Fusion genes are a class of oncogenes that can be targeted for therapeutics or as diagnostic tools because of their inherent expression in tumor tissues. Next-generation sequencing has revolutionized sequencing by creating high-throughput and low-cost methods, and fusion gene detection has become easy and cost-effective. Targeting of fusion genes has facilitated the development of several successful anti-cancer drugs and targeted therapies [53]. The roles of lncRNAs in genomic regulation are multiple, they involve transcriptional and post-transcriptional regulation, and imprinting and epigenetic modifications. Studies have documented lncRNAs that play roles in tumorigenesis and tumor progression [15–17]; in particular, aberrant lncRNA expression or lncRNA dysfunction can lead to DNA damage, immune escape, and cellular metabolic disorders in cancer cells. lncRNAs can regulate cell proliferation, apoptosis, migration, invasion, and maintenance of stemness during the development of cancer. In addition, many lncRNAs are transcriptionally regulated by key tumor suppressors or oncogenes [15]. The inherent heterogeneity and diversity of the lncRNA world make the complex tumorigenesis process more interesting than it would otherwise be [17]. Hence, identification and characterization of lncRNAs could lead to development of new clinical approaches to understand complex tumorigenesis and to treat cancers. In this study, we identified and characterized fusion genes, lncRNAs, and miRNAs from RNA-seq data of thymoma patients, identified aberrant lncRNAs and miRNAs in thymomas, built an lncRNA-mRNA-miRNA regulatory network in thymoma, and performed a functional enrichment analysis.
Our RNA-seq analysis results show frequent translocations of fusion gene events with KMT2A-MAML2, and others such as HADHB-REEP1, COQ3-CGA, and MCM4-SNTB1. KMT2A encodes a transcriptional co-factor that plays an important role in gene expression regulation during early development and hematopoiesis. This protein binds DNA and methylates the histone H3 at lysine-4 to regulate HOX and other genes. KMT2A has been reported to be a translocation partner with more than 80 unique fusion partners [54]. MAML2, a member of the Mastermind-like family of proteins, binds the ankyrin repeat domain Notch receptors (ICN1-4). The KMT2A-MAML2 gene fusion was reported in a patient with therapy-related acute myeloid leukemia [55], and it was also reported in aggressive histologic thymoma subtypes [56], a finding in line with our results. The KMT2A-MAML2 fusion has also been shown to suppresses promoter activation of the NOTCH1 target gene HES1 [57], and it demonstrates oncogenic activities [58]. The next most frequent fusion genes detected in our analysis were HADHB-REEP1 and COQ3-CGA. HADHB encodes a mitochondrial trifunctional protein associated with peroxisomal disease, valproic acid pathway, and mitochondrial fatty acid beta-oxidation. REEP1 encodes a mitochondrial protein whose mutation has been reported to cause spastic paraplegia, which is a neurodegenerative disorder. To the best of our knowledge, these 2 fusions are novel, and have not been reported before. The gene fusion MCM4-SNTB1 was reported in a study including a single case of metastatic thymic adenocarcinoma [59]. Other fusion transcripts reported in thymomas are FABP2-C4orf3 and CTBS-GNG5 [59], but no fusion events in TETs have been published [5]. Although, gene GTF2I is the most frequently mutated and is considered a master genetic regulator in thymomas; it has not been found to be involved in fusion events [5,60,61].
To our astonishment, we detected differentially-expressed lncRNAs (DEMs), many reported in the literature as promoting the proliferation of various malignancies including gastric, ovarian, breast, prostate cancers, non-small cell lung cancer (NSCLC), leukemia, sarcomas, and head and neck squamous cell carcinomas [62–71]. LINC01485 has been shown to be responsible for the formation of gastric cancer by suppression of EGFR ubiquitination and triggering of the Akt signaling pathway [70]. AFAP1-AS1 has also been shown to cause severe human cancers [67]. LINC01697 is involved in the development of lung squamous cell carcinoma and can be used as a biomarker [72].
Another lncRNA, LINC00324, has been shown to cause gastric cancer by interacting with the human antigen R (HuR) [73]. ADAMTS9-AS1 has been proved to inhibit the Wnt signaling pathway in colorectal cancer, and can be considered a prognostic marker [74]. VLDLR-AS1 has been suggested to promote fat loss in cancer cachexia [75]. On the other hand, NEAT1 has been shown to promote a variety of human cancers [68], while LINC00968 has been recognized as an oncogene that activates the cancer prevalent signaling pathways; mainly Akt, PI3K, and mTOR pathways [76]. The lncRNAs
Our study detected several DEMs, most of them upregulated in thymoma tissue and associated with different biological and disease pathways. For instance, the hsa-let-7a family of miRNAs inhibit migration, invasion, and tumor growth by targeting AKT2 in papillary thyroid carcinoma [48]. Our network and pathway analysis of differentially-expressed lncRNA-mRNA-miRNA showed that overexpression of miRNA clusters activated the PI3K-Akt/FoxO/HIF-1/Rap-1 signal pathway, suggesting a possible role for PI3K/Akt/HIF-1/Rap-1 inhibitors in thymoma patients.
Conclusions
In this study, we reported fusion genes, lncRNAs, and miRNAs detected from RNA-seq data of thymoma patients. Also, we detected aberrant lncRNAs and miRNAs, constructed a lncRNA-mRNA-miRNA regulatory network, and performed a functional enrichment analysis. We detected 21 fusion genes, out of which 5 were significantly expressed and unique in thymoma tissues (KMT2A-MAML2, HADHB-REEP1, COQ3-CGA, MCM4-SNTB1, and IFT140-ACTN4); some of them have been reported in the literature. Additionally, we reported 65 differentially-expressed lncRNAs in thymomas, including AFAP1-AS1, LINC00324, ADAMTS9-AS1, VLDLR-AS1, LINC00968, and NEAT1, that have been validated with the TCGA database. Our DFS analysis suggests that markers with both low or high expressions directly affect patient survival. Our DFS map demonstrated that aberrant expression of these lncRNAs can be used to differentiate thymomas from other cancers. The functional annotation of the top 10 ranked lncRNAs was associated with specific molecular functions.
Our analysis of small ncRNAs detected 1695 miRNAs, with a significant number of miRNAs overexpressed in thymomas. Network analysis of lncRNA-mRNA-miRNA regulation identified a group of miRNAs upregulated in thymomas and triggering the expression of target protein-coding genes. As a result, many biological pathways are disrupted, including the PI3K-Akt signaling pathway, the FoxO signaling pathway, the HIF-1 signaling pathway, and proteoglycans. Finally, our gene set enrichment analysis revealed that these target protein-coding genes are associated with various molecular functions, including systemic immune response, hepatic immune response, positive regulation of DNA replication, positive regulation of cell proliferation, positive regulation of MAPK cascade, negative regulation of extrinsic apoptotic signaling pathway, and negative regulation of apoptosis. Our results show that overexpression of miRNA clusters activates the PI3K-Akt, FoxO, HIF-1, and Rap-1 signaling pathways, suggesting that PI3K, Akt, HIF-1, and Rap-1 inhibitors may be therapeutic in patients with thymoma.
Figures
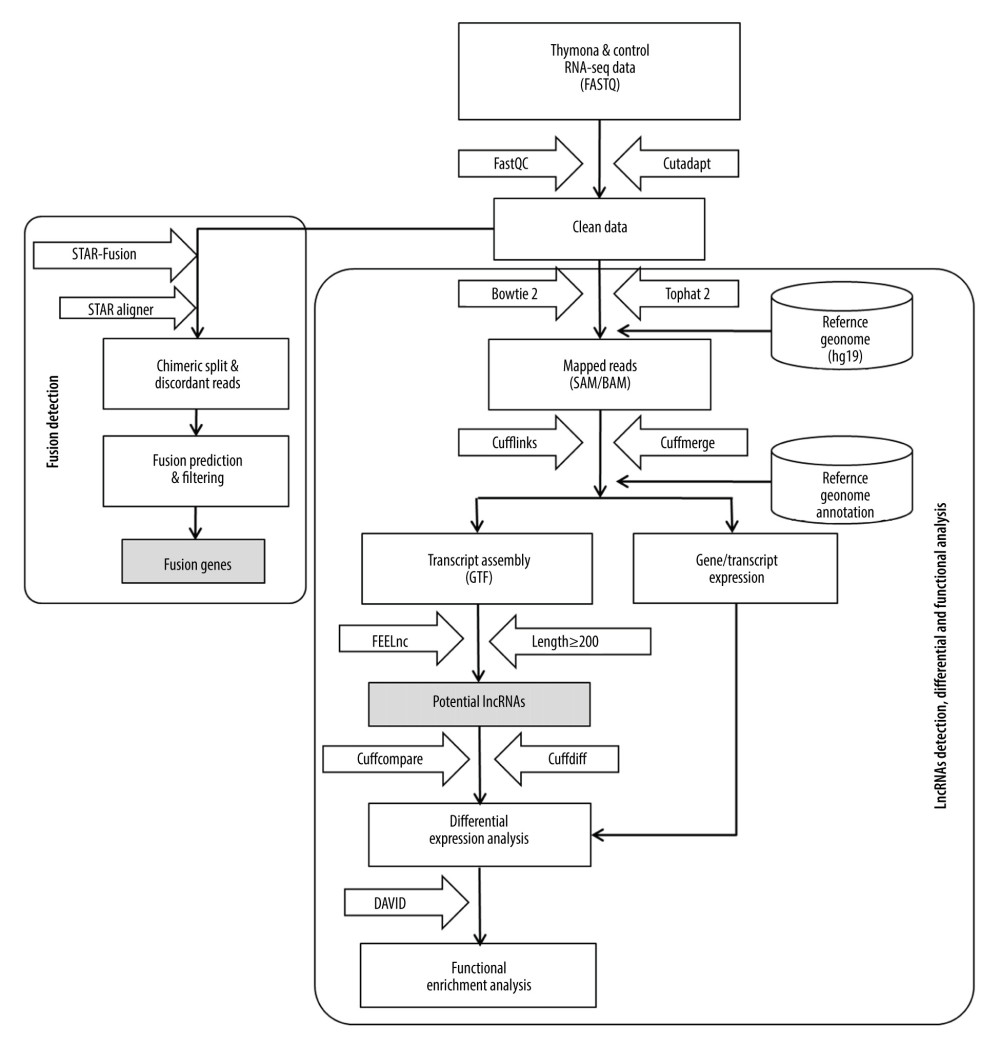 Figure 1. Methodological pipeline to process RNA-Seq data and analysis used in this study.
Figure 1. Methodological pipeline to process RNA-Seq data and analysis used in this study. 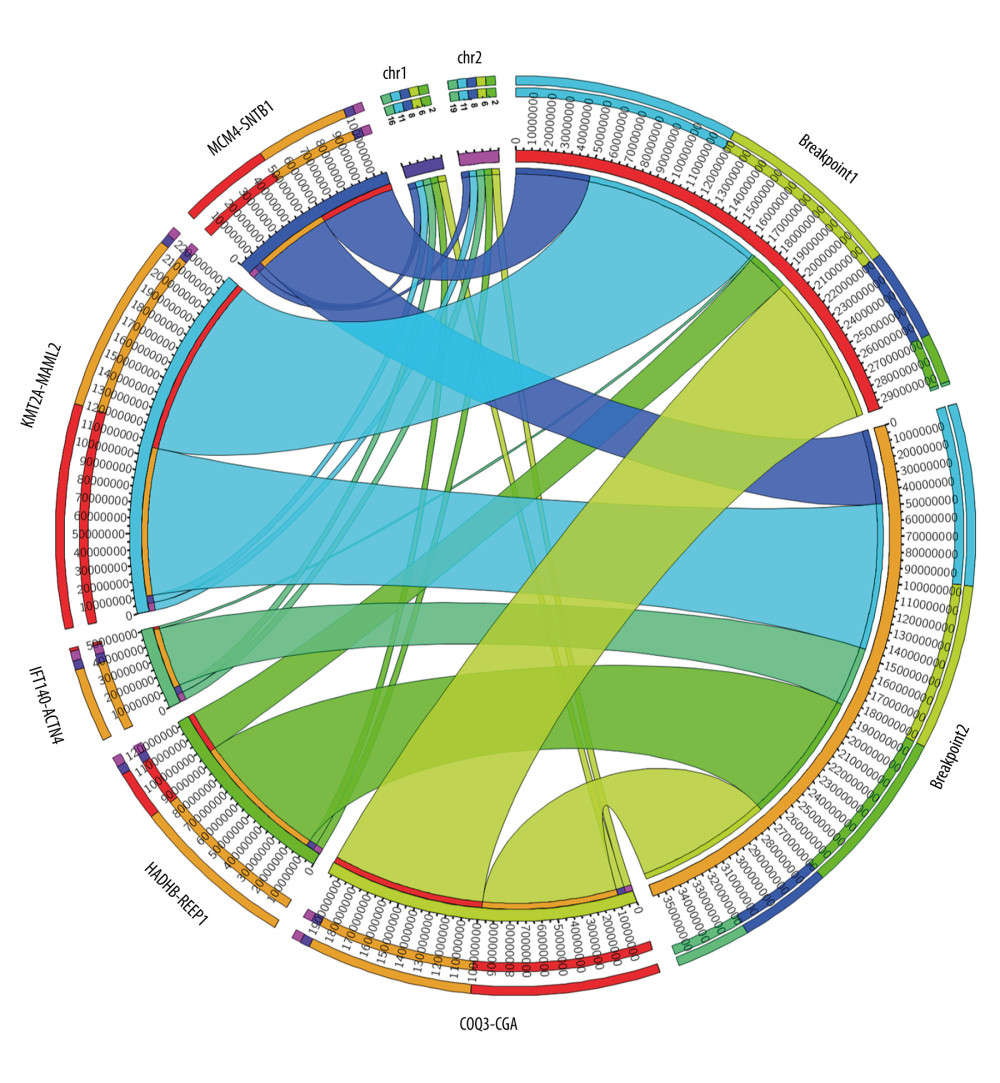 Figure 2. Circos plot of fusion genes mapped to chromosome and breakpoints on genome.
Figure 2. Circos plot of fusion genes mapped to chromosome and breakpoints on genome. ![(A–F) Boxplot of normalized expression [log2(TPM+1)] of thymoma samples (red color) and normal samples (gray color), and disease-free survival (DFS) analysis of patients in high- (red line) and low-expression (blue line) groups for the top 6 dysregulated lncRNAs.](https://jours.isi-science.com/imageXml.php?i=medscimonit-27-e929727-g003.jpg&idArt=929727&w=1000) Figure 3. (A–F) Boxplot of normalized expression [log2(TPM+1)] of thymoma samples (red color) and normal samples (gray color), and disease-free survival (DFS) analysis of patients in high- (red line) and low-expression (blue line) groups for the top 6 dysregulated lncRNAs.
Figure 3. (A–F) Boxplot of normalized expression [log2(TPM+1)] of thymoma samples (red color) and normal samples (gray color), and disease-free survival (DFS) analysis of patients in high- (red line) and low-expression (blue line) groups for the top 6 dysregulated lncRNAs. 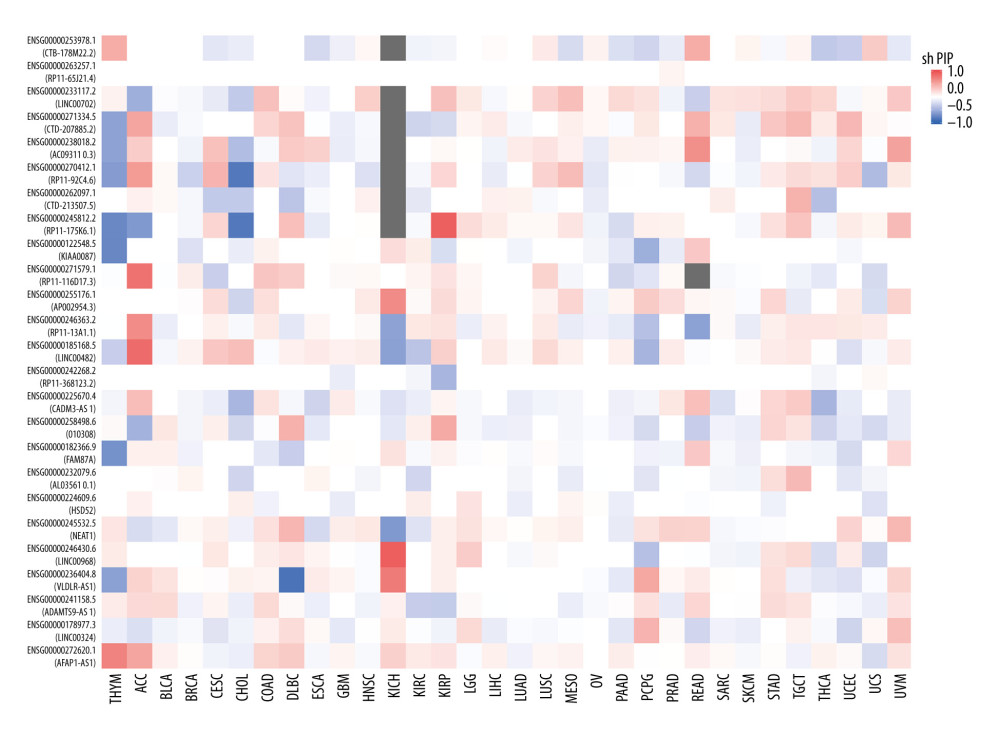 Figure 4. Disease-free survival (DFS) map showing survival contribution of top 25 dysregulated lncRNAs in various cancers including thymoma (THYM) using TCGA datasets with a significance level of 0.05 estimated using the Mantel-Cox test.
Figure 4. Disease-free survival (DFS) map showing survival contribution of top 25 dysregulated lncRNAs in various cancers including thymoma (THYM) using TCGA datasets with a significance level of 0.05 estimated using the Mantel-Cox test. 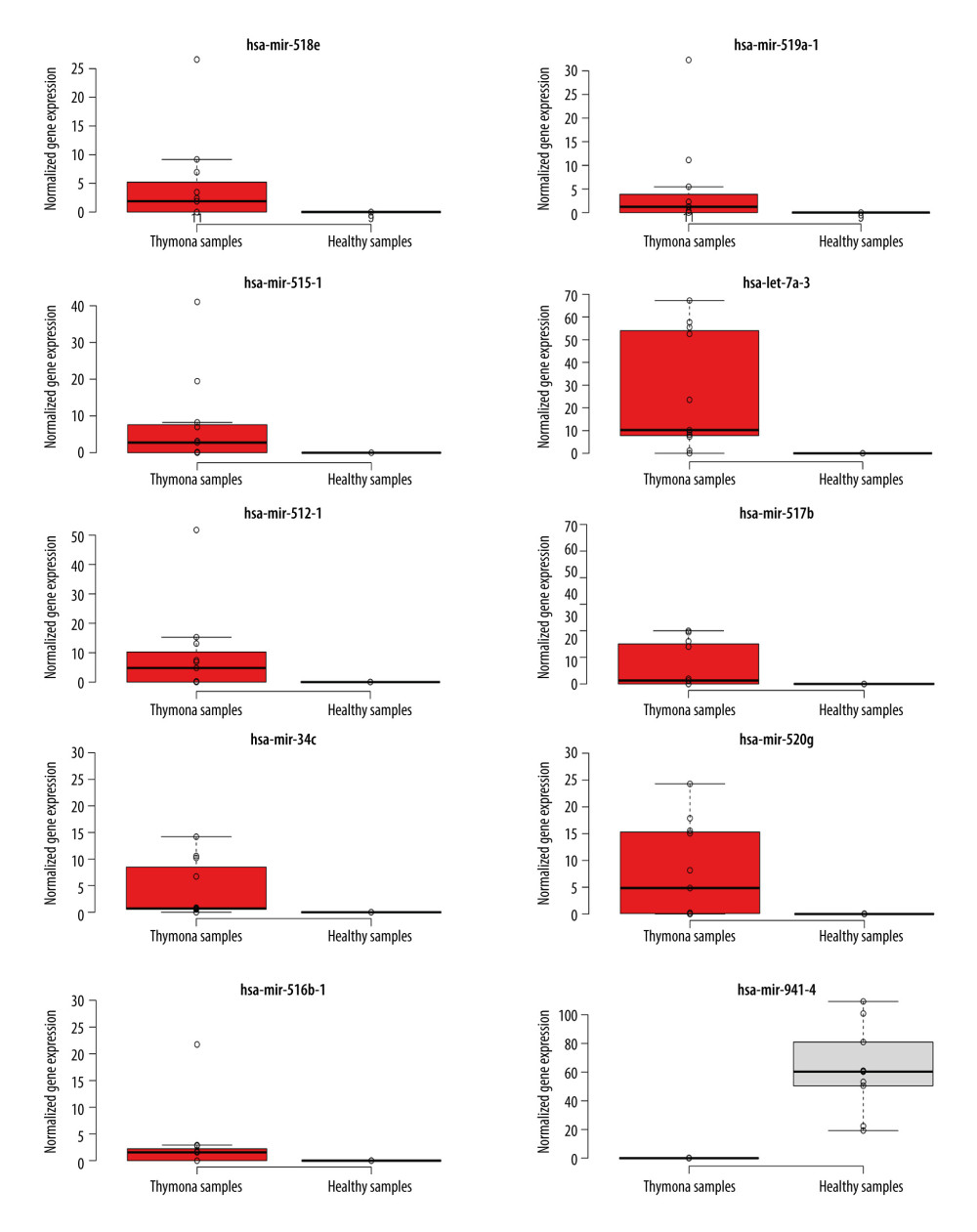 Figure 5. Boxplot of first 10 differentially-expressed miRNAs (DEMs) showing their normalized gene expression over thymoma versus those of healthy samples.
Figure 5. Boxplot of first 10 differentially-expressed miRNAs (DEMs) showing their normalized gene expression over thymoma versus those of healthy samples. 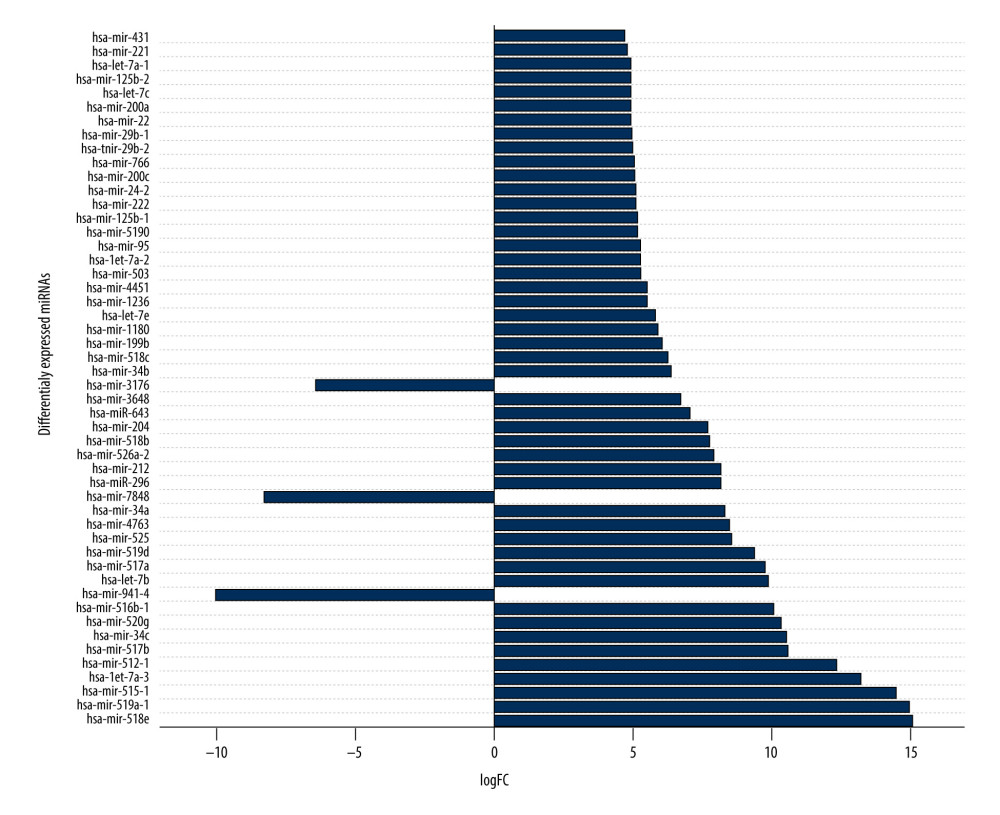 Figure 6. First 50 differentially-expressed miRNAs (DEMs) and their logFC values.
Figure 6. First 50 differentially-expressed miRNAs (DEMs) and their logFC values. 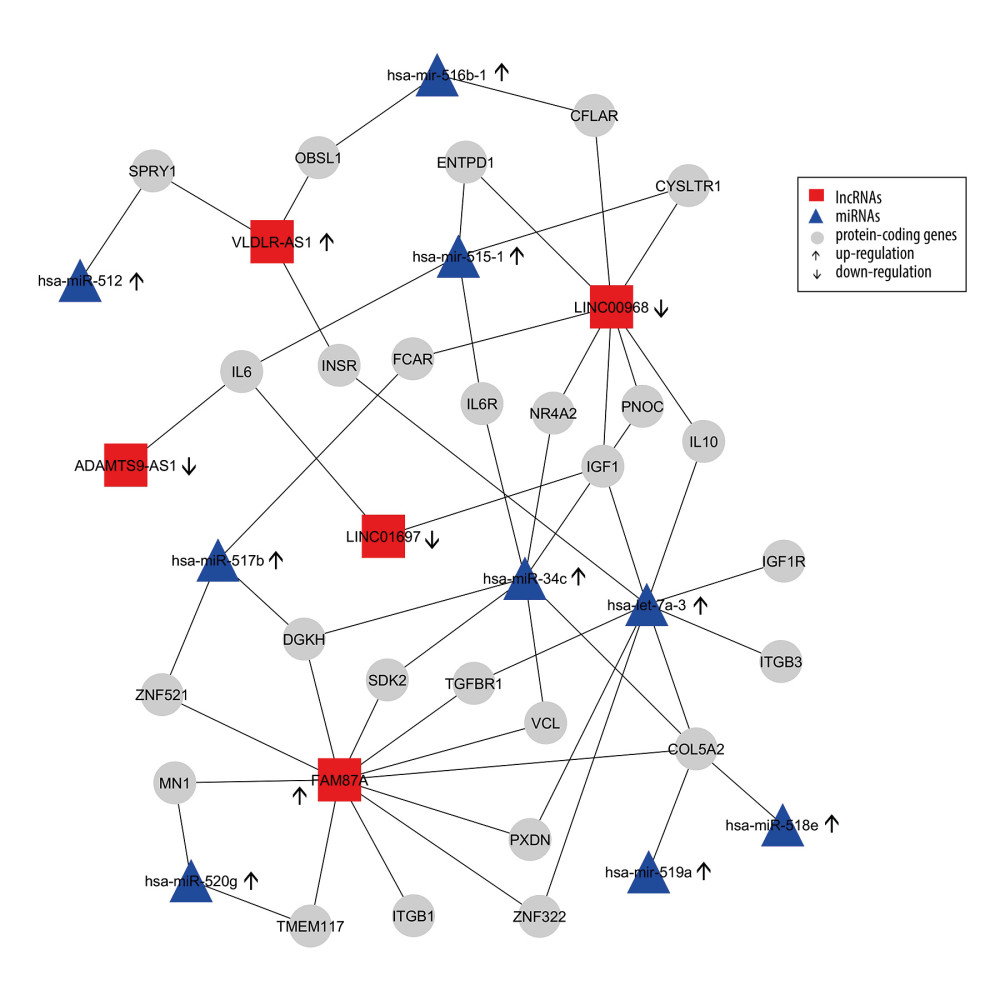 Figure 7. Differentially-expressed lncRNA-mRNA-miRNA interaction network in thymoma. Boxes represent lncRNAs, circles represent coding genes, and triangles represent miRNAs. Up arrows represent upregulated nodes, while down arrows represent downregulated nodes in thymomas. Lines between lncRNAs-mRNAs-miRNAs represent regulatory networks among them.
Figure 7. Differentially-expressed lncRNA-mRNA-miRNA interaction network in thymoma. Boxes represent lncRNAs, circles represent coding genes, and triangles represent miRNAs. Up arrows represent upregulated nodes, while down arrows represent downregulated nodes in thymomas. Lines between lncRNAs-mRNAs-miRNAs represent regulatory networks among them. Tables
Table 1. Basic characteristics of considered small RNA-seq data. Table 2. Characteristics of study samples of thymus tumor/cancer (n=25) and healthy thymus. (n=25) with the number of missing values.
Table 2. Characteristics of study samples of thymus tumor/cancer (n=25) and healthy thymus. (n=25) with the number of missing values. Table 3. Summary of detected fusion genes and their statistics (arranged by significance level).
Table 3. Summary of detected fusion genes and their statistics (arranged by significance level).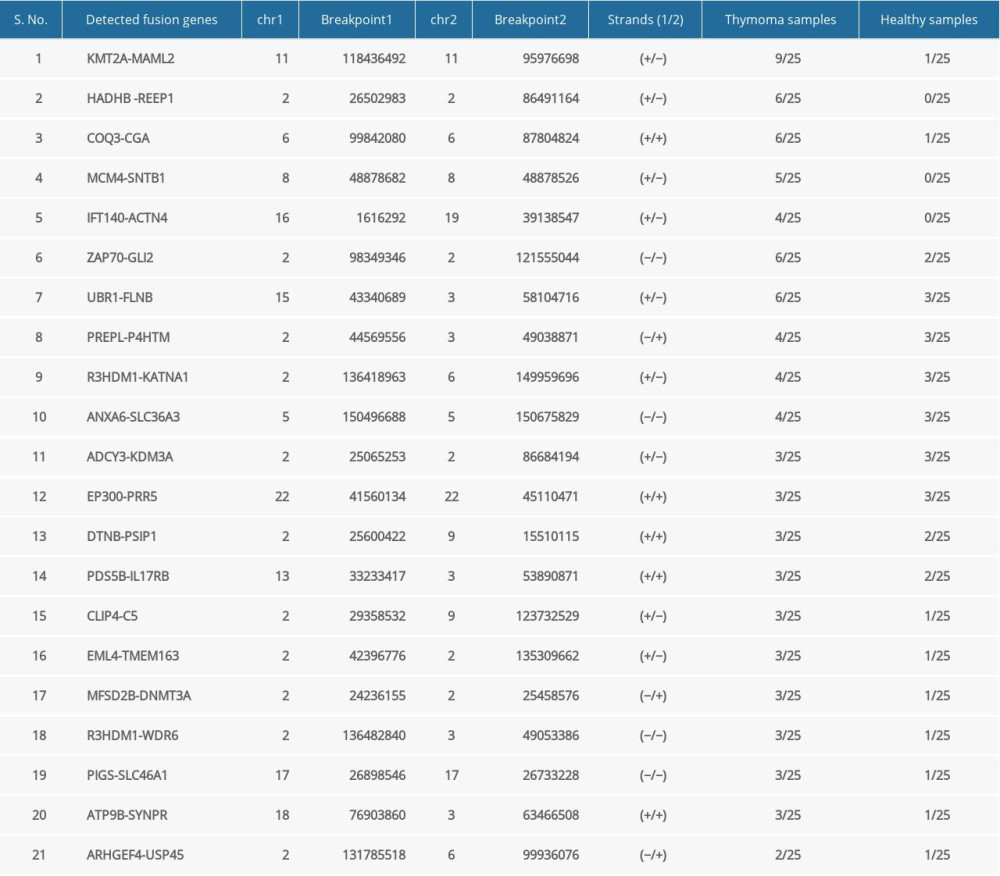 Table 4. lncRNAs dysregulated in thymoma discovered by RNA-seq and validated by TCGA.
Table 4. lncRNAs dysregulated in thymoma discovered by RNA-seq and validated by TCGA.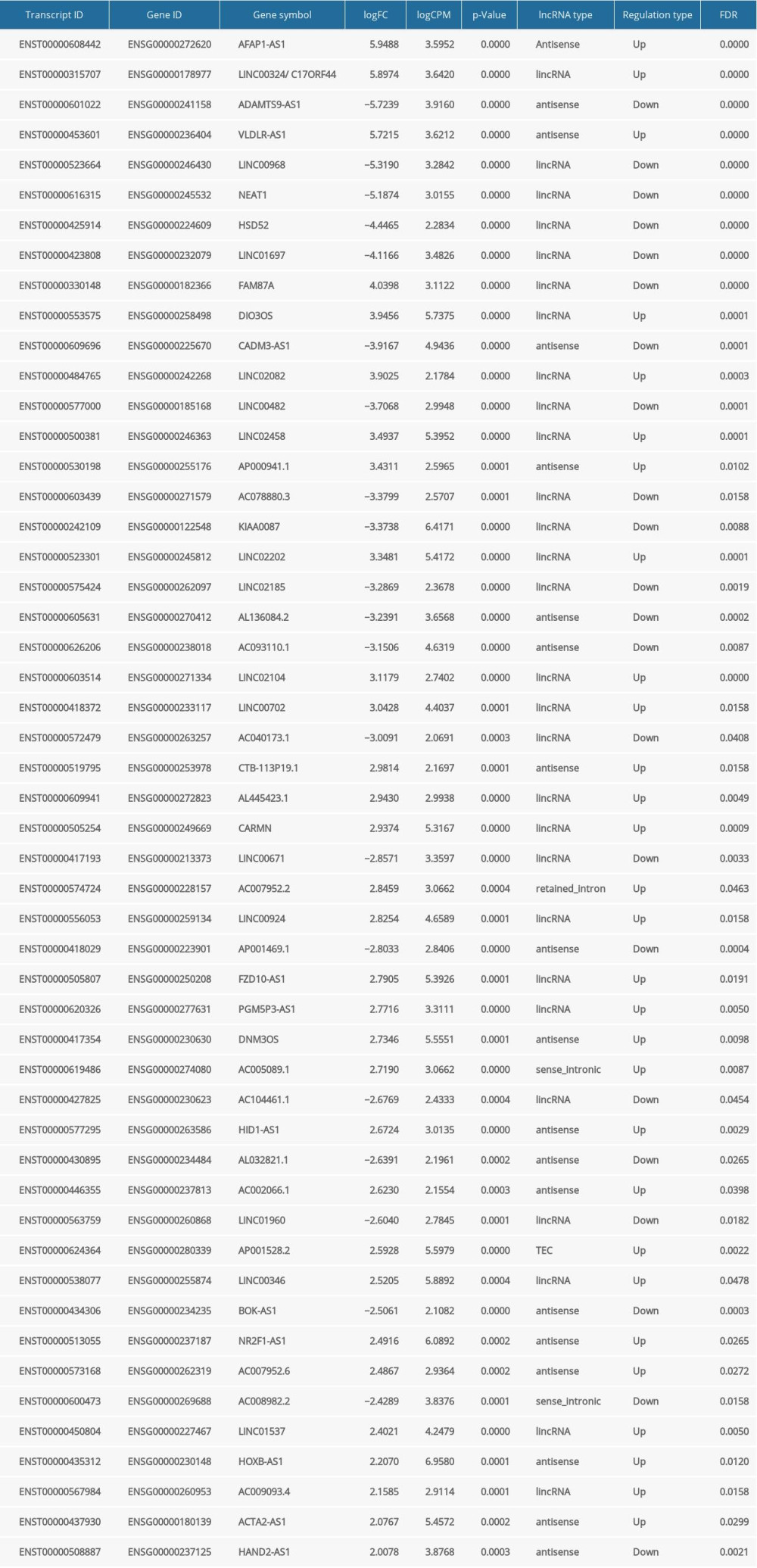 Table 5. lncRNAs and their predicted protein interactions using the UCSC-TFBS algorithm.
Table 5. lncRNAs and their predicted protein interactions using the UCSC-TFBS algorithm.
References
1. Chaudhry MS, Velardi E, Dudakov JA, Thymus: The next (re)generation: Immunol Rev, 2016; 271; 56-71
2. Eng TY, Fuller CD, Jagirdar J, Thymic carcinoma: State of the art review: Int J Radiat Oncol Biol Phys, 2004; 59; 654-64
3. Zucali PA, De Pas T, Palmieri G, Phase II study of everolimus in patients with thymoma and thymic carcinoma previously treated with cisplatin-based chemotherapy: J Clin Oncol, 2018; 36; 342-49
4. Zhao C, Rajan A, Immune checkpoint inhibitors for treatment of thymic epithelial tumors: how to maximize benefit and optimize risk?: Mediastinum, 2019; 3; 35
5. Radovich M, Pickering CR, Felau I, The integrated genomic landscape of thymic epithelial tumors: Cancer Cell, 2018; 33; 244-58.e10
6. Rashid MA, Newton MAH, Hoque MdT, Mixing energy models in genetic algorithms for on-lattice protein structure prediction: Biomed Res Int, 2013; 2013; 924137
7. Marx A, Chan JKC, Coindre J-M, The 2015 World Health Organization classification of tumors of the thymus: Continuity and changes: J Thorac Oncol, 2015; 10; 1383-95
8. Meng F-J, Wang S, Zhang J, Alteration in gene expression profiles of thymoma: Genetic differences and potential novel targets: Thorac Cancer, 2019; 10; 1129-35
9. Detterbeck FC, Parsons AM, Thymic tumors: Ann Thorac Surg, 2004; 77; 1860-69
10. Srirajaskanthan R, Toubanakis C, Dusmet M, A review of thymic tumours: Lung Cancer, 2008; 60; 4-13
11. Mangan KF, Volkin R, Winkelstein A, Autoreactive erythroid progenitor-T suppressor cells in the pure red cell aplasia associated with thymoma and panhypogammaglobulinemia: Am J Hematol, 1986; 23; 167-73
12. Fried AJ, Bonilla FA, Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections: Clin Microbiol Rev, 2009; 22; 396-414
13. ENCODE Project Consortium, The ENCODE (ENCyclopedia Of DNA Elements) project: Science, 2004; 306; 636-40
14. Da Sacco L, Baldassarre A, Masotti A, Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis: Int J Mol Sci, 2011; 13; 97-114
15. Huarte M, The emerging role of lncRNAs in cancer: Nat Med, 2015; 21; 1253-61
16. Schmitt AM, Chang HY, Long noncoding RNAs in cancer pathways: Cancer Cell, 2016; 29; 452-63
17. Jiang M-C, Ni J-J, Cui W-Y, Emerging roles of lncRNA in cancer and therapeutic opportunities: Am J Cancer Res, 2019; 9; 1354-66
18. Yu Y-P, Liu P, Nelson J, Identification of recurrent fusion genes across multiple cancer types: Sci Rep, 2019; 9; 1074
19. Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads: EMBnet.journal, 2011; 17; 10-12
20. Robinson DR, Wu Y-M, Kalyana-Sundaram S, Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing: Nat Genet, 2013; 45; 180-85
21. Tate JG, Bamford S, Jubb HC, COSMIC: The Catalogue Of Somatic Mutations In Cancer: Nucleic Acids Res, 2019; 47; D941-47
22. Lee SJ, Hong JY, Kim K, Detection of fusion genes using a targeted RNA sequencing panel in gastrointestinal and rare cancers: J Oncol, 2020; 2020; 4659062
23. Fernandez-Cuesta L, Sun R, Menon R, Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data: Genome Biol, 2015; 16; 7
24. Cleynen A, Szalat R, Kemal Samur M, Expressed fusion gene landscape and its impact in multiple myeloma: Nat Commun, 2017; 8; 1893
25. Schram AM, Chang MT, Jonsson P, Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance: Nat Rev Clin Oncol, 2017; 14; 735-48
26. McPherson A, Hormozdiari F, Zayed A, deFuse: An algorithm for gene fusion discovery in tumor RNA-Seq data: PLoS Comput Biol, 2011; 7; e1001138
27. Benelli M, Pescucci C, Marseglia G, Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript: Bioinformatics, 2012; 28; 3232-39
28. Davidson NM, Majewski IJ, Oshlack A, JAFFA: High sensitivity transcriptome-focused fusion gene detection: Genome Med, 2015; 7; 43
29. Haas BJ, Dobin A, Stransky N, STAR-Fusion: Fast and accurate fusion transcript detection from RNA-Seq: bioRxiv, 2017; 2017; 120295
30. Ma C, Shao M, Kingsford C, SQUID: Transcriptomic structural variation detection from RNA-seq: Genome Biol, 2018; 19; 52
31. Haas BJ, Dobin A, Li B, Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods: Genome Biol, 2019; 20; 213
32. Dobin A, Davis CA, Schlesinger F, STAR: Ultrafast universal RNA-seq aligner: Bioinformatics, 2013; 29; 15-21
33. Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2: Nat Methods, 2012; 9; 357-59
34. Kim D, Pertea G, Trapnell C, Pimentel H, TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions: Genome Biol, 2013; 14; R36
35. Trapnell C, Roberts A, Goff L, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks: Nat Protoc, 2012; 7; 562-78
36. Trapnell C, Hendrickson DG, Sauvageau M, Differential analysis of gene regulation at transcript resolution with RNA-seq: Nat Biotechnol, 2013; 31; 46-53
37. Wucher V, Legeai F, Hédan B, FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome: Nucleic Acids Res, 2017; 45; e57
38. Faiza M, Tanveer K, Fatihi S, Comprehensive overview and assessment of miRNA target prediction tools in human and drosophila melanogaster: Current Bioinformatics, 2017; 14; 432-45
39. Lewis BP, Burge CB, Bartel DP, Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets: Cell, 2005; 120; 15-20
40. Stuopelytė K, Daniūnaitė K, Jankevičius F, Detection of miRNAs in urine of prostate cancer patients: Medicina (Kaunas), 2016; 52; 116-24
41. Lu J, Getz G, Miska EA, MicroRNA expression profiles classify human cancers: Nature, 2005; 435; 834-38
42. An J, Lai J, Lehman M, MiRDeep*: An integrated application tool for miRNA identification from RNA sequencing data: Nucleic Acids Res, 2012; 41; 727-37
43. Chen Y, Wang X, miRDB: An online database for prediction of functional microRNA targets: Nucleic Acids Res, 2020; 48; D127-31
44. Raza K, Analysis of microarray data using artificial intelligence-based techniques: Biotechnology, 2019; 865-88
45. Raza K, Reconstruction, topological and gene ontology enrichment analysis of cancerous gene regulatory network modules: Current Bioinformatics, 2016; 11; 1-1
46. Krzywinski M, Schein J, Birol I, Circos: An information aesthetic for comparative genomics: Genome Res, 2009; 19; 1639-45
47. Singh S, Li H, Prediction, characterization, and in silico validation of chimeric RNAs: Methods Mol Biol, 2020; 2079; 3-12
48. Zhou B, Shan H, Su Y, Let-7a inhibits migration, invasion and tumor growth by targeting AKT2 in papillary thyroid carcinoma: Oncotarget, 2017; 8; 69746-55
49. Chirshev E, Oberg KC, Ioffe YJ, Let-7 as biomarker, prognostic indicator, and therapy for precision medicine in cancer: Clin Transl Med, 2019; 8; 24
50. ASCO, Thymoma and thymic carcinoma – symptoms and signs: CancerNet, 2012
51. Engels EA, Epidemiology of thymoma and associated malignancies: J Thorac Oncol, 2010; 5; S260-65
52. Liu A, Gao X, Zhao L, Thymoma with acute gastric volvulus: A case report: BMC Cancer, 2017; 17; 801
53. Kamps R, Brandão RD, van den Bosch BJ, Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification: Int J Mol Sci, 2017; 18; 308
54. Milne TA, Briggs SD, Brock HW, MLL targets SET domain methyltransferase activity to Hox gene promoters: Mol Cell, 2002; 10; 1107-17
55. Obama K, Furukawa Y, Tara M, Secondary monocytic leukemia with rearrangement of the MLL gene occurring during the course of adult T-cell leukemia: Int J Hematol, 1998; 68; 323-26
56. Massoth LR, Hung YP, Dias-Santagata D, Pan-cancer landscape analysis reveals recurrent KMT2A-MAML2 gene fusion in aggressive histologic subtypes of thymoma: JCO Precis Oncol, 2020; 4; PO.19.00288
57. Nemoto N, Suzukawa K, Shimizu S, Identification of a novel fusion gene MLL-MAML2 in secondary acute myelogenous leukemia and myelodysplastic syndrome with inv(11)(q21q23): Genes Chromosomes Cancer, 2007; 46; 813-19
58. Wächter K, Kowarz E, Marschalek R, Functional characterisation of different MLL fusion proteins by using inducible Sleeping Beauty vectors: Cancer Lett, 2014; 352; 196-202
59. Lee Y, Park S, Lee S-H, Lee H, Characterization of genetic aberrations in a single case of metastatic thymic adenocarcinoma: BMC Cancer, 2017; 17; 330
60. Petrini I, Meltzer PS, Kim I-K, A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors: Nat Genet, 2014; 46; 844-49
61. Oberndorfer F, Müllauer L, Genomic alterations in thymoma – molecular pathogenesis?: J Thoracic Dis, 2020; 12; 7536-44
62. Guo S, Chen W, Luo Y, Clinical implication of long non-coding RNA NEAT1 expression in hepatocellular carcinoma patients: Int J Clin Exp Pathol, 2015; 8; 5395-402
63. Han D, Wang J, Cheng G, LncRNA NEAT1 enhances the radio-resistance of cervical cancer via miR-193b-3p/CCND1 axis: Oncotarget, 2018; 9; 2395-409
64. Huang G, He X, Wei X-L, lncRNA NEAT1 promotes cell proliferation and invasion by regulating miR-365/RGS20 in oral squamous cell carcinoma: Oncol Rep, 2018; 39; 1948-56
65. Li X, Ren Y, Zuo T, Long noncoding RNA LINC00978 promotes cell proliferation and invasion in non-small cell lung cancer by inhibiting miR-6754-5p: Mol Med Rep, 2018; 18; 4725-32
66. Fang X-N, Yin M, Li H, Comprehensive analysis of competitive endogenous RNAs network associated with head and neck squamous cell carcinoma: Sci Rep, 2018; 8; 10544
67. Ji D, Zhong X, Jiang X, The role of long non-coding RNA AFAP1-AS1 in human malignant tumors: Pathol Res Pract, 2018; 214; 1524-31
68. Dong P, Xiong Y, Yue J, Long non-coding RNA NEAT1: A novel target for diagnosis and therapy in human tumors: Front Genet, 2018; 9; 471
69. Xiu D-H, Liu G-F, Yu S-N, Long non-coding RNA LINC00968 attenuates drug resistance of breast cancer cells through inhibiting the Wnt2/β-catenin signaling pathway by regulating WNT2: J Exp Clin Cancer Res, 2019; 38; 94
70. Zhou J, Wu L, Li W, Long noncoding RNA LINC01485 promotes tumor growth and migration via inhibiting EGFR ubiquitination and activating EGFR/Akt signaling in gastric cancer: Onco Targets Ther, 2020; 13; 8413-25
71. Li Y, Cao X, Li H, Identification and validation of novel long non-coding RNA biomarkers for early diagnosis of oral squamous cell carcinoma: Front Bioeng Biotechnol, 2020; 8; 256
72. Liu J, Yao Y, Hu Z, Transcriptional profiling of long-intergenic noncoding RNAs in lung squamous cell carcinoma and its value in diagnosis and prognosis: Mol Genet Genomic Med, 2019; 7; e994
73. Zou Z, Ma T, He X, Long intergenic non-coding RNA 00324 promotes gastric cancer cell proliferation via binding with HuR and stabilizing FAM83B expression: Cell Death Dis, 2018; 9; 717
74. Li N, Li J, Mi Q, Long non-coding RNA ADAMTS9-AS1 suppresses colorectal cancer by inhibiting the Wnt/β-catenin signalling pathway and is a potential diagnostic biomarker: J Cell Mol Med, 2020; 24; 11318-29
75. Liu H, Zhou T, Wang B, Identification and functional analysis of a potential key lncRNA involved in fat loss of cancer cachexia: J Cell Biochem, 2018; 119; 1679-88
76. Liu G, Yuan D, Sun P, LINC00968 functions as an oncogene in osteosarcoma by activating the PI3K/AKT/mTOR signaling: J Cell Physiol, 2018; 233; 8639-47
77. Gong J, Jin S, Pan X, Identification of long non-coding RNAs for predicting prognosis among patients with thymoma: Clin Lab, 2018; 64; 1193-98
78. Wan J-F, Wan J-Y, Dong C, Linc00324 promotes the progression of papillary thyroid cancer via regulating Notch signaling pathway: Eur Rev Med Pharmacol Sci, 2020; 24; 6818-24
79. Huang EY, Gallegos AM, Richards SM, Surface expression of Notch1 on thymocytes: Correlation with the double-negative to double-positive transition: J Immunol, 2003; 171; 2296-304
80. Ye L, Jin W, Identification of lncRNA-associated competing endogenous RNA networks for occurrence and prognosis of gastric carcinoma, 2020 rs-45596/v1
81. Su Y, Chen Y, Tian Z, lncRNAs classifier to accurately predict the recurrence of thymic epithelial tumors: Thorac Cancer, 2020; 11; 1773-83
82. Luo Y-Y, Wang Z-H, Yu Q, LncRNA-NEAT1 promotes proliferation of T-ALL cells via miR-146b-5p/NOTCH1 signaling pathway: Pathol Res Pract, 2020; 216; 153212
Figures
Figure 1. Methodological pipeline to process RNA-Seq data and analysis used in this study.Figure 2. Circos plot of fusion genes mapped to chromosome and breakpoints on genome.Figure 3. (A–F) Boxplot of normalized expression [log2(TPM+1)] of thymoma samples (red color) and normal samples (gray color), and disease-free survival (DFS) analysis of patients in high- (red line) and low-expression (blue line) groups for the top 6 dysregulated lncRNAs.Figure 4. Disease-free survival (DFS) map showing survival contribution of top 25 dysregulated lncRNAs in various cancers including thymoma (THYM) using TCGA datasets with a significance level of 0.05 estimated using the Mantel-Cox test.Figure 5. Boxplot of first 10 differentially-expressed miRNAs (DEMs) showing their normalized gene expression over thymoma versus those of healthy samples.Figure 6. First 50 differentially-expressed miRNAs (DEMs) and their logFC values.Figure 7. Differentially-expressed lncRNA-mRNA-miRNA interaction network in thymoma. Boxes represent lncRNAs, circles represent coding genes, and triangles represent miRNAs. Up arrows represent upregulated nodes, while down arrows represent downregulated nodes in thymomas. Lines between lncRNAs-mRNAs-miRNAs represent regulatory networks among them. Tables
Table 1. Basic characteristics of considered small RNA-seq data.Table 2. Characteristics of study samples of thymus tumor/cancer (n=25) and healthy thymus. (n=25) with the number of missing values.Table 3. Summary of detected fusion genes and their statistics (arranged by significance level).Table 4. lncRNAs dysregulated in thymoma discovered by RNA-seq and validated by TCGA.Table 5. lncRNAs and their predicted protein interactions using the UCSC-TFBS algorithm.Table 1. Basic characteristics of considered small RNA-seq data.Table 2. Characteristics of study samples of thymus tumor/cancer (n=25) and healthy thymus. (n=25) with the number of missing values.Table 3. Summary of detected fusion genes and their statistics (arranged by significance level).Table 4. lncRNAs dysregulated in thymoma discovered by RNA-seq and validated by TCGA.Table 5. lncRNAs and their predicted protein interactions using the UCSC-TFBS algorithm. In Press
12 Mar 2024 : Clinical Research
Metabolomic Alterations in Methotrexate Treatment of Moderate-to-Severe PsoriasisMed Sci Monit In Press; DOI: 10.12659/MSM.943360
14 Mar 2024 : Clinical Research
Renal Dysfunction Increases Risk of Adverse Cardiovascular Events in 5-Year Follow-Up Study of Intermediate...Med Sci Monit In Press; DOI: 10.12659/MSM.943956
15 Mar 2024 : Clinical Research
Impact of One-Lung Ventilation on Oxygenation and Ventilation Time in Thoracoscopic Heart Surgery: A Compar...Med Sci Monit In Press; DOI: 10.12659/MSM.943089
14 Mar 2024 : Clinical Research
Differential DHA and EPA Levels in Women with Preterm and Term Births: A Tertiary Hospital Study in IndonesiaMed Sci Monit In Press; DOI: 10.12659/MSM.943895
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952