18 May 2021: Database Analysis
Identification of a DNA Methylation-Based Prognostic Signature for Patients with Triple-Negative Breast Cancer
Yinqi Gao1ABCE, Xuelong Wang2AC, Shihui Li1CE, Zhiqiang Zhang1CE, Xuefei Li1CE, Fangcai Lin1AE*DOI: 10.12659/MSM.930025
Med Sci Monit 2021; 27:e930025






Abstract
BACKGROUND: Aberrant DNA methylation is an important biological regulatory mechanism in malignant tumors. However, it remains underutilized for establishing prognostic models for triple-negative breast cancer (TNBC).
MATERIAL AND METHODS: Methylation data and expression data downloaded from The Cancer Genome Atlas (TCGA) were used to identify differentially methylated sites (DMSs). The prognosis-related DMSs were selected by univariate Cox regression analysis. Functional enrichment was analyzed using DAVID. A protein-protein interaction (PPI) network was constructed using STRING. Finally, a methylation-based prognostic signature was constructed using LASSO method and further validated in 2 validation cohorts.
RESULTS: Firstly, we identified 743 DMSs corresponding to 332 genes, including 357 hypermethylated sites and 386 hypomethylated sites. Furthermore, we selected 103 prognosis-related DMSs by univariate Cox regression. Using a LASSO algorithm, we established a 5-DMSs prognostic signature in TCGA-TNBC cohort, which could classify TNBC patients with significant survival difference (log-rank p=4.97E-03). Patients in the high-risk group had shorter overall survival than patients in the low-risk group. The excellent performance was validated in GSE78754 (HR=2.42, 95%CI: 1.27-4.59, log-rank P=0.0055). Moreover, for disease-free survival, the prognostic performance was verified in GSE141441 (HR=2.09, 95%CI: 1.28-3.44, log-rank P=0.0027). Multivariate Cox regression analysis indicated that the 5-DMSs signature could serve as an independent risk factor.
CONCLUSIONS: We constructed a 5-DMSs signature with excellent performance for the prediction of disease-free survival and overall survival, providing a guide for clinicians in directing personalized therapeutic regimen selection of TNBC patients.
Keywords: DNA Methylation, Triple Negative Breast Neoplasms, Aged, 80 and over, Biomarkers, Tumor, Cohort Studies, Disease-Free Survival, Protein Interaction Maps
Background
Breast cancer (BC) is a common malignant tumor that occurs in the thymic epithelial tissue, and it has become the main cause of mortality and morbidity in women [1,2]. Triple-negative breast cancer (TNBC), defined as the loss of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER2) expression, is a particularly aggressive subtype of BC, accounting for 15–20% of all breast cancers [3]. Compared to other types of BC, patients with TNBC usually have poorer overall survival (OS) and a higher probability of cancer recurrence due to the lack of early detection biomarkers and clear therapeutic targets [4]. Hence, it is urgent to develop robust prognostic biomarkers to guide personalized therapy.
DNA methylation is one of the most commonly occurring epigenetic events that regulate gene expression without altering the DNA sequence [5]. Generally, cytosine residues in the CpGs, frequently found in the proximal promoter regions of many genes, are methylated by DNA methyltransferases that catalyze the transfer of the active methyl group from S-adenosylmethionine to the C5-position of cytosine [6–8]. It has been shown that hypermethylation at promoter CpG islands typically results in decreased transcription of downstream genes [9]. When methylation is experimentally removed from promoter regions, transcription levels rise [10]. Increasing studies have revealed that epigenetic gene silencing caused by DNA methylation could play important roles in tumorigenesis and progression [11–13]. It has also been found that the aberrant DNA methylation could be an important risk factor in TNBC [14,15]. For example, Prajzendanc et al found that BRCA1 promoter methylation in peripheral blood cells was frequently detected in TNBC using methylation-sensitive high-resolution melting, which was significantly associated with reduced BRCA1 expression, aggressive phenotype, and poor prognosis [16]. BRCA1 hypermethylation confers a homologous recombination deficiency, immune cell type, genome-wide DNA methylation, and transcriptional phenotype similar to TNBC tumors with BRCA1-inactivating variants. Given that DNA methylation changes are plausibly critical components of the molecular mechanisms involved in TNBC, distinct DNA methylation could be a potential biomarker to improve the accuracy of TNBC diagnosis and prognosis. In addition, the emergence of high-throughput technology makes it possible to identify reliable biomarkers for diagnosis and prognosis. Several studies have proposed many DNA methylation signatures to improve the accuracy of cancer prognosis [17–19]. However, few robust DNA methylation biomarkers were identified as available indicators. Although a nomogram incorporating a prognostic risk model and clinicopathological features was developed by Peng et al for predicting the prognosis of TNBC [20], more reliable methylation-based biomarkers to predict the survival are needed for personalized therapy of TNBC patients.
In the present study, based on the DNA methylation data downloaded from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO), we identified differentially methylated sites (DMSs) with consistent difference. Combining gene expression data and clinical data of TNBC samples downloaded from TCGA, we further screened the prognosis-related DMSs with aberrant gene expression. Based on prognosis-related DMSs, we aimed to establish a prognostic signature to classify TNBC patients into different risk groups. The prognostic performance was validated in 2 independent cohorts. Such a signature could provide effective help for prognosis classification and personalized therapy of TNBC patients.
Material and Methods
DATA COLLECTION:
TNBC samples and normal breast samples with methylation data (Platform: Illumina Infinium HumanMethylation450), mRNA-Seq data (Platform: Illumina HiSeq 2000 RNA sequencing), and clinical information were downloaded from TCGA (https://gdc-portal.nci.nih.gov/). Patients with unknown survival time, age, tumor stage, and lymph node metastasis status were excluded. Ultimately, 114 TNBC samples and 86 normal breast samples were retained in our study. A DNA methylation cohort (Platform: Illumina Infinium HumanMethylation450) was derived from the GEO database, which contains 14 TNBC samples and 17 normal breast samples. Moreover, 2 DNA methylation cohorts (Platform: Illumina Infinium HumanMethylation450) with clinical information (GSE141441 and GSE78754), containing 130 and 63 TNBC samples, respectively, were downloaded from the GEO database as the validation cohorts. All cohorts analyzed in the present study are included in Table 1. Approval by the Institutional Ethics Committee was not necessary because all data were collected from the publicly available GEO and TCGA databases.
SCREENING OF DIFFERENTIALLY METHYLATED SITES:
The DNA methylation data, defined as β values, were derived from the GEO and TCGA databases. The methylation data processed by background subtraction, quantile normalization, and quality control had normal distribution. Thus, the
FUNCTIONAL ENRICHMENT ANALYSIS:
Function enrichment, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, was performed for DEGs corresponding to DMSs using the Database for Annotation, Visualization and Integrated Discovery (DAVID,
PROTEIN–PROTEIN INTERACTION (PPI) NETWORK ANALYSIS:
The PPI network provides a meaningful insight into the molecular mechanisms of key cellular activities in cancer progression. Based on DEGs containing DMSs, a PPI network was established using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (
CONSTRUCTION OF PROGNOSTIC SIGNATURE:
Using univariate Cox regression analysis, DMSs significantly associated with overall survival (OS) were identified with a
Coefsite denote the coefficient of a prognosis-related DMS. Methylationsite is the methylation level of the same prognosis-related DMS. A risk score was calculated for each patient using the prognostic model. Then, patients were classified into a high-risk group and a low-risk group based on the median risk score.
STATISTICAL ANALYSIS:
The
Results
PATIENT CHARACTERISTICS:
A total of 114 patients collected from TCGA were included as the training cohort for constructing the prognostic signature. In the training cohort, most patients had stage II disease (64.7%), and only 2 patients had stage IV disease. Seventy-one (62.3%) patients had lymph node metastasis. In the validation cohort GSE141441, most patients also had stage II disease (55.4%), and no patients with stage IV were included. Furthermore, more than half of patients had recurrence. In the validation cohort GSE78754, tumor stage was not recorded and the vital status of most patients was death. The clinicopathological characteristics of the patients are summarized in Table 2.
IDENTIFICATION OF DIFFERENTIALLY METHYLATED SITES IN TNBC:
With cutoff criteria of FDR <0.05, 250 024 and 180 148 DMSs were selected from TCGA-TNBC and GSE52865 cohorts, respectively. As shown in Figure 1A, a total of 110 228 DMSs, including 49 199 hypermethylation sites and 61 029 hypomethylation sites, were selected with 95.9% concordance score (binomial test, P<0.0001). Moreover, we identified 1049 DEGs with cutoff criteria of FDR <0.05 and |logFC| >2 (Figure 1B), including 568 upregulated genes and 481 downregulated genes. To identify DMSs that directly regulate gene expression, we calculated the Pearson correlation coefficient to observe the association between the methylation value of a DMS and the expression value of the corresponding DEG. A total of 743 DMSs, whose methylation levels were significantly negatively associated with expression levels of 332 corresponding DEGs, were selected for further analysis.
FUNCTIONAL ENRICHMENT:
Functional enrichment analysis was performed using DAVID. The significant terms enriched from each GO category are shown in Figure 2A. The results showed that several cancer-related terms were significantly enriched. The most significant GO terms were angiogenesis (P<0.001). Extracellular matrix organization, inflammatory response, and cell adhesion were also significantly enriched. As shown in Figure 2B, KEGG analysis suggested that the DEGs were significantly enriched in cytokine-cytokine receptor interaction, PI3K-Akt signaling pathway, and ECM-receptor interaction. Functional enrichment analysis indicated that DMSs affected the occurrence and progression of TNBC by regulating the corresponding gene expression.
CONSTRUCTION OF PPI NETWORK:
To further explore the interactions between the 332 DEGs, a PPI network was constructed. With an interaction score >0.7 as the cutoff criterion, a PPI network containing 168 nodes and 396 edges was constructed using STRING (Figure 3A). Furthermore, as shown in Figure 3B, 10 hub genes with high degrees of connectivity within the PPI network were identified using the cytoHubba plugin for Cytoscape, including kinesin family member 2C (KIF2C), polo-like kinase 1 (PLK1), centromere protein F (CENPF), baculoviral IAP repeat containing 5 (BIRC5), mitotic arrest deficient 2-like 1 (MAD2L1), protein regulator of cytokinesis 1 (PRC1), ribonucleotide reductase regulatory subunit M2 (RRM2), forkhead box M1 (FOXM1), NIMA-related kinase 2 (NEK2), and kinesin family member C1 (KIFC1).
ESTABLISHMENT OF A 5-DMSS PROGNOSTIC SIGNATURE:
A total of 103 prognosis-related DMSs, including 46 hypermethylation sites and 57 hypomethylation sites, were identified by univariate Cox proportional hazards regression analysis (P <0.05). Supplementary Table 1 contains a list of 103 DMSs. Based on these prognosis-related DMSs, we established a 5-DMSs signature to guide the prediction of prognosis for TNBC patients using the LASSO algorithm. The features of 5-DMSs are shown in Table 3. Thus, the prognostic scoring model was constructed as follows: Risk score=(−1.664× methylation degree of cg15724876)+(−0.373× methylation degree of cg17887364)+(−0.249× methylation degree of cg19419246)+(0.753× methylation degree of cg21234506)+(0.149× methylation degree of cg21580376). Two of the 5-DMSs were related to high risk (cg21234506 and cg21580376; HR >1), while 3 of the 5-DMSs appeared to be protective (cg15724876, cg17887364, and cg19419246; HR <1). We calculated the risk scores for patients in TCGA-TNBC cohort and then classified them into high-risk and low-risk groups using the median risk score as the cutoff. We found that patients in the high-risk group had poorer OS than those in the low-risk group (log-rank P=4.97E-03, Figure 4A). Multivariate Cox regression analysis showed that the 5-DMSs signature could serve as an independent prognostic factor (HR=7.55, 95% CI=1.39–40.9, P=0.019, Figure 4B). The distributions of the risk scores, survival time, survival status, and 5-DMSs methylation profiles are shown in Figure 4C. We also performed the stratification analysis for stage to prove an independent prediction of the 5-DMSs prognostic signature. For 92 patients with stage I or II, 47 patients were classified into the low-risk group, and the rest were classified into the high-risk group. Kaplan-Meier survival curve analysis showed that patients in the high-risk group had significantly shorter OS (log-rank P=0.015, Figure 5A). Of the 22 patients with stage III or IV, 10 patients were classified into the low-risk group, and the rest were classified into the high-risk group. Although there was no statistically significant survival difference between the 2 risk groups, it still had a trend of poor prognosis in the high-risk group (log-rank P=0.248, Figure 5B). The methylation differences of the 5-DMSs between high-risk and low-risk groups are shown in Figure 6A. The results showed that patients in the low-risk group had significantly higher methylation degrees of 3 protective DMSs, while patients in the high-risk group had significantly higher methylation degrees of 1 risky DMS. Although another risky DMS was not statistically significant, it still had a trend of high methylation degree in the high-risk group.
PERFORMANCE VALIDATION OF THE 5-DMSS PROGNOSTIC SIGNATURE:
Using the 5-DMSs prognostic signature, each patient in GSE78754 was classified into a high-risk (n=32) or a low-risk (n=31) group. Kaplan-Meier survival curve analysis showed that high-risk scores were significantly correlated with poor prognosis (log-rank P=0.0055, Figure 7B). Multivariate Cox regression analysis also confirmed the independent prognostic performance of the 5-DMSs signature (HR=2.66, 95%CI=1.38–5.11, P=0.003, Table 4). Similar to findings in the training cohort, the methylation differences of each DMS were observed (Figure 6C). Moreover, another independent cohort (GSE141441) containing 130 patients with disease-free survival data was used to validate the prognostic performance for tumor recurrence. The results showed that patients in the low-risk group had a longer disease-free survival (log-rank P=0.0027, Figure 7A). Multivariate Cox regression analysis suggested that the 5-DMSs signature could serve as an independent prognostic factor for disease-free survival (HR=1.77, 95%CI=1.05–3.00, P=0.033, Table 4). The stratification analysis for stage also proved an independent prediction of the 5-DMSs prognostic signature. For 116 patients with stage I or II, 65 patients were classified into the low-risk group and the rest were classified into the high-risk group. Kaplan-Meier survival curve analysis showed that patients in the high-risk group had significantly shorter survival than those in the low-risk group (log-rank p=1.48E-03, Figure 5C). Moreover, all patients with stage III or IV were classified into the high-risk group. Observing the methylation differences of each DMS, we found that patients in the high-risk group tended to have higher methylation degrees at 2 risky sites, whereas patients in the low-risk group tended to have higher methylation degrees at 3 protective sites (Figure 6B). The risk scores distributions, survival time, survival status, and 5-DMSs methylation profiles for patients in GSE141441 and GSE78754 cohorts are shown in Figure 7C and 7D, respectively.
Discussion
TNBC is the most fatal subtype of breast cancer, which is a heterogeneous group of tumors with one common feature: a distinctly aggressive nature with higher rates of relapse and shorter overall survival in the metastatic setting [21,22]. DNA methylation refers to heritable and modifiable markers that regulate gene expression without changing the DNA sequence, which could be a potential biomarker to improve the accuracy of TNBC prognosis. In the present study, which comprehensively analyzed methylation data, expression data, and clinical data from TCGA, we identified prognosis-related DMSs and further developed a novel 5-DMSs prognostic signature, which had improved ability to predict prognosis for TNBC patients compared with clinical risk factors. Multivariate Cox regression analysis showed that the 5-DMSs signature could serve as an independent prognostic factor to categorize TNBC patients with different outcomes. The prognostic performance for overall survival and disease-free survival was validated in independent cohorts. Our study provides a reliable prognostic signature for TNBC patients based on DNA methylation, and also identified potential therapeutic targets.
Functional enrichment analysis provided an intuitive overview of the mechanism of TNBC. The most significant GO terms were angiogenesis, a core hallmark of advanced cancer. Many studies have demonstrated that angiogenesis is important in the transformation of hyperplastic in situ epithelium to invasive carcinoma for TNBC growth and metastasis [23–25]. Other significantly enriched GO terms, such as extracellular matrix organization, inflammatory response, and cell adhesion, are also closely associated with TNBC metastasis and chemoresistance [26–28]. KEGG pathway analysis showed significant enrichment of cytokine-cytokine receptor interaction and PI3K-Akt signaling pathway. Cytokine-cytokine receptor interaction is an important way to regulate cell growth and proliferation and participate in immune response and inflammation, especially in tumor growth and metastasis [29–31]. The PI3K-Akt signaling pathway is an important and active pathway involved in chemoresistance and survival, which is speculated to play an important part in malignant transformation and has been considered as a potential molecular target for the design of therapeutic agents to treat TNBC [32–34]. These findings indicate that the identified DMSs affect the occurrence and progression of TNBC by regulating the corresponding gene expression.
We constructed a PPI network to investigate the functional interactions between DEGs and identified 10 hub genes with the highest degree of connectivity from the PPI network. Some of these genes have been reported to be closely associated with TNBC, such as CENPF, PLK1, and KIF2C. Sun et al found that CENPF could promote BC bone metastasis by activating PI3K-AKT-mTORC1 signaling and might be a novel therapeutic target for BC treatment [35]. Ueda et al found that PLK1 was significantly overexpressed in TNBC tissues and plays a pivotal role in the regulation of mitosis of TNBC cells, which could be an attractive molecular-targeted therapy for TNBC [36].
In our study, based on LASSO regression analysis, we developed a novel prognostic signature based on 5 methylation sites (cg15724876, cg17887364, cg19419246, cg21234506, and cg21580376) corresponding to 5 genes: transforming growth factor beta receptor 2 (TGFBR2), eukaryotic translation initiation factor 4E binding protein 1 (EIF4EBP1), FBJ murine osteosarcoma viral oncogene homolog B (FOSB), BCL2-related protein A1 (BCL2A1), and adrenoceptor beta 2 (ADRB2). Cox regression analysis showed that 3 of 5 sites (cg15724876, cg17887364, and cg19419246) were protective for TNBC, and their methylation degrees were significantly higher in the low-risk group classified by prognostic signature. Moreover, the other 2 sites were significantly hypermethylated in the high-risk group as risky factors. Although few studies have reported the biological mechanism of 5 methylation sites in tumor progression, the epigenetic changes of several corresponding genes have been reported to be related to carcinogenesis, migration, and invasion. Zhang et al reported that EZH2-mediated histone 3 trimethylated on lysine 27 (H3K27me3), a marker of silent chromatin conformation, at the FOSB promoter, inhibited its expression. EZH2 inhibitor promotes the shift from H3K27me3 to H3K27ac at the FOSB promoter, and recruits the transcription factor C/EBPβ to activate FOSB gene transcription. Epigenetic inactivation of FOSB mediated by EZH2 increased the gene expression in TNBC samples and further promoted TNBC cells proliferation, indicating a critical role of FOSB methylation in the regulation of TNBC progression [37]. Ma et al reported that TGFBR2, an important tumor suppressor mediating TGF-β signaling and inducing cell cycle arrest, is downregulated in esophageal squamous cell carcinoma due to DNA hypermethylation of its promoter regions, and is involved in malignant transformation of esophageal squamous cell carcinoma by inducing cell cycle G2/M arrest [38]. All 5 genes have been reported to be involved in many cancer-related pathways to regulate TNBC progression [39–43]. For instance, Karlsson et al found that EIF4EBP1 and S6K2 have a correlated mRNA expression, and that high levels of EIF4EBP1 and S6K2 were associated with a poor prognosis, independent of other classical prognostic markers [44]. In our study, we found that EIF4EBP1 at cg17887364 was hypomethylated in TNBC, which might regulate the gene overexpression and further induce poor prognosis. However, no previous studies have reported on the association between abnormal methylation of TGFBR2, EIF4EBP1, BCL2A1, or ADRB2 and TNBC. Hence, it is necessary to perform further studies on the methylation regulation mechanism of these genes and TNBC.
Cancer is a complex regulatory network in which multisite methylation as biomarkers could achieve higher specificity and sensitivity compared with single-site methylation. Peng et al [20] developed a 15-CpG-based signature for predicting prognosis in TNBC based on a TCGA-TNBC cohort to make a prognosis classification for TNBC patients. Unfortunately, the insufficient external validation limited it as a clinically reliable indicator. Our 5-DMSs signature was carefully developed with a particular focus on this issue. In our study, using the combination of weighted methylation values of these 5-DMSs, we constructed a 5-DMSs signature for the prediction of prognosis and validated the prognostic performance with a larger sample size containing 2 external cohorts. In methodology, we further evaluated the association between methylation levels of DMSs and expression levels of the corresponding DEGs, allowing us to screen for more meaningful DMSs related to tumor progression. However, limitations still exist in this study. First, although this study was validated using GEO data, prospective studies and multicenter clinical trials are needed to for further clinical validation for the 5-DMSs prognostic signature. Second, lack of mechanism studies hinders better application of the 5-DMSs prognostic signature. Therefore, experimental research on each DMS within the 5-DMSs prognostic signature should be performed, which might provide significant information to further the understanding of their functional roles.
Conclusions
In summary, comprehensively analyzing the methylation data, expression data, and clinical data, we constructed a potential prognostic tool, named the 5-DMSs signature, to predict prognosis for patients with TNBC, which might provide support for clinicians in directing personalized therapeutic regimen selection.
Figures
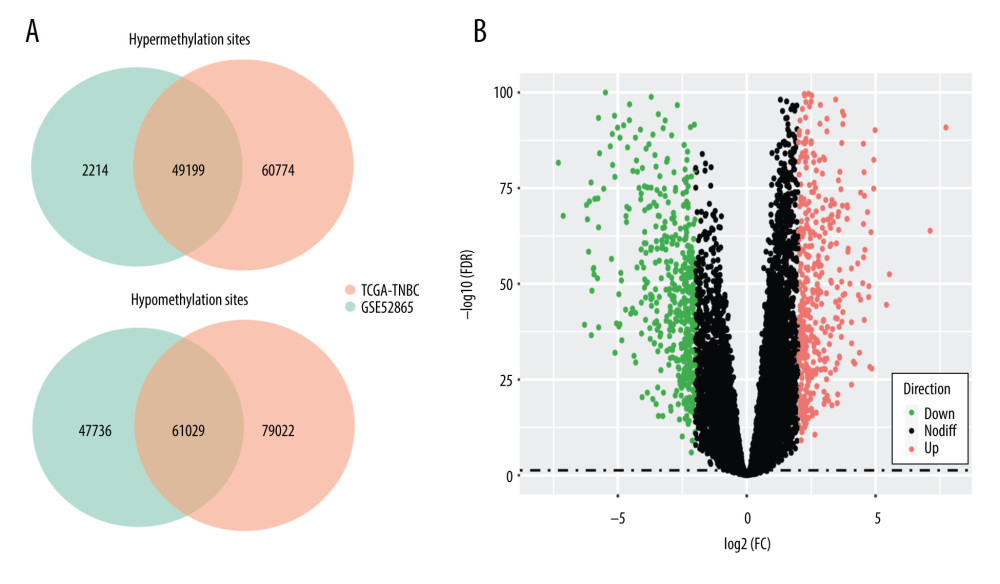 Figure 1. Consistency of differentially methylated sites and identification of differentially expressed genes. (A) Overlapping differentially methylated sites between TCGA-TNBC and GSE52865. (B) Volcano plot of differentially expressed genes.
Figure 1. Consistency of differentially methylated sites and identification of differentially expressed genes. (A) Overlapping differentially methylated sites between TCGA-TNBC and GSE52865. (B) Volcano plot of differentially expressed genes. 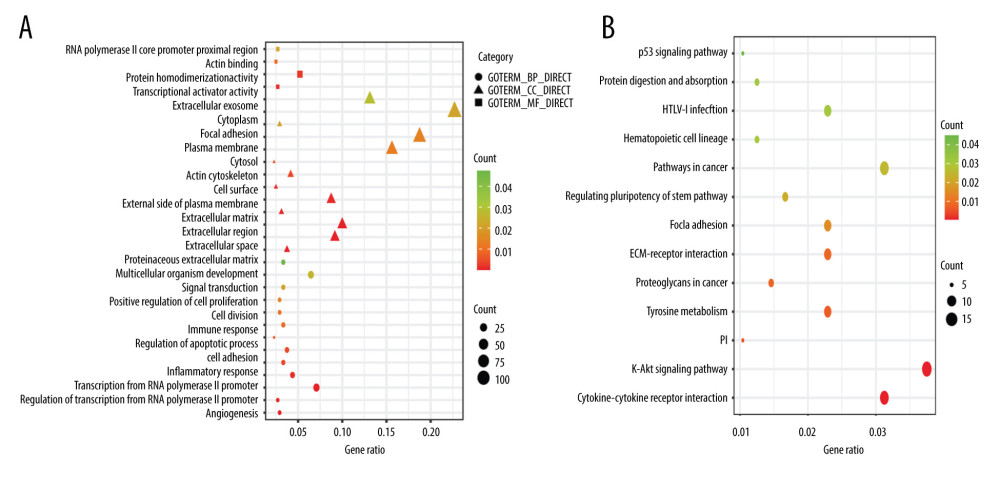 Figure 2. Functional enrichment of the significantly enriched Gene Ontology terms (A) and Kyoto Encyclopedia of Genes and Genomes pathways (B) of differentially expressed genes containing differentially methylated sites.
Figure 2. Functional enrichment of the significantly enriched Gene Ontology terms (A) and Kyoto Encyclopedia of Genes and Genomes pathways (B) of differentially expressed genes containing differentially methylated sites. 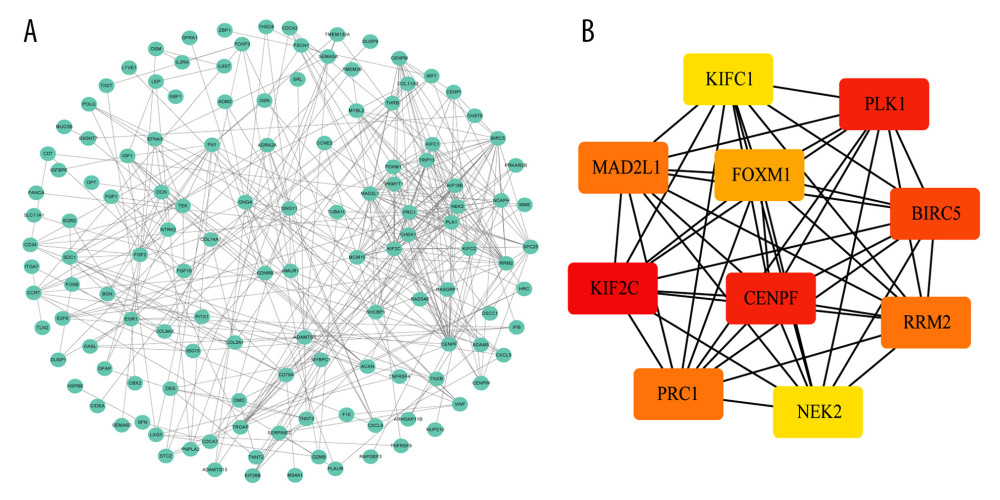 Figure 3. Identification of hub genes within protein-protein interaction (PPI) network. (A) PPI network. (B) Top 10 hub genes with high degrees of connectivity.
Figure 3. Identification of hub genes within protein-protein interaction (PPI) network. (A) PPI network. (B) Top 10 hub genes with high degrees of connectivity. 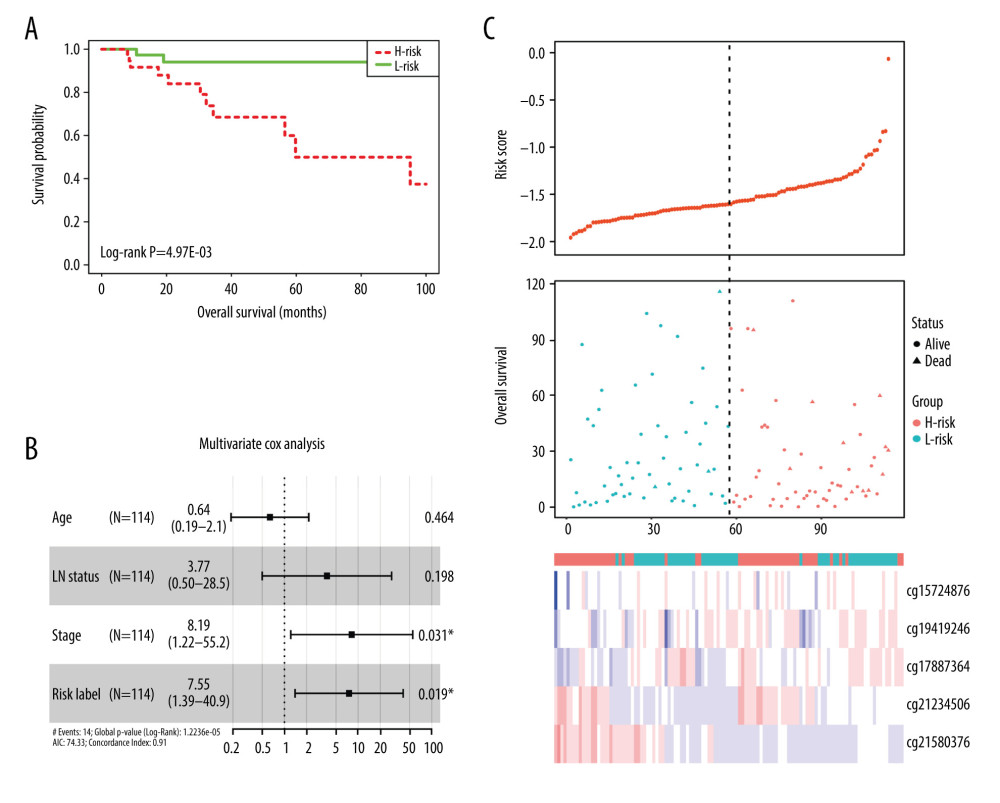 Figure 4. Evaluating the prognostic power of the 5-DMSs signature in the TCGA-TNBC cohort. (A) Kaplan-Meier survival curve of the high- and low-risk groups. (B) Multivariate analysis of risk factors for TNBC. (C) Distribution of risk score, survival status, and methylation heatmap of the 5-DMSs.
Figure 4. Evaluating the prognostic power of the 5-DMSs signature in the TCGA-TNBC cohort. (A) Kaplan-Meier survival curve of the high- and low-risk groups. (B) Multivariate analysis of risk factors for TNBC. (C) Distribution of risk score, survival status, and methylation heatmap of the 5-DMSs. 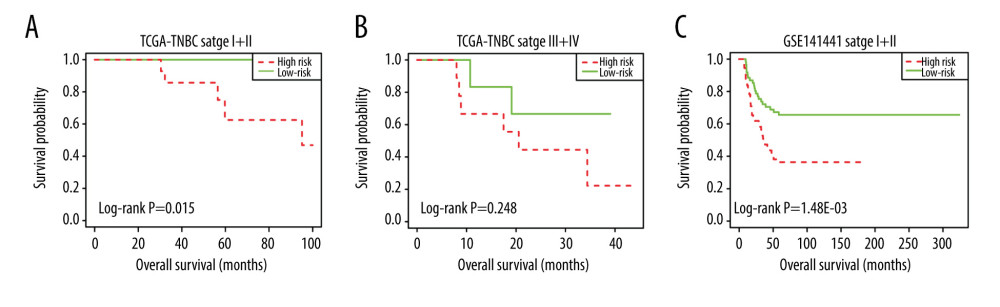 Figure 5. The stratification analysis for stage in TCGA-TNBC (A) and GSE141441 (B).
Figure 5. The stratification analysis for stage in TCGA-TNBC (A) and GSE141441 (B). 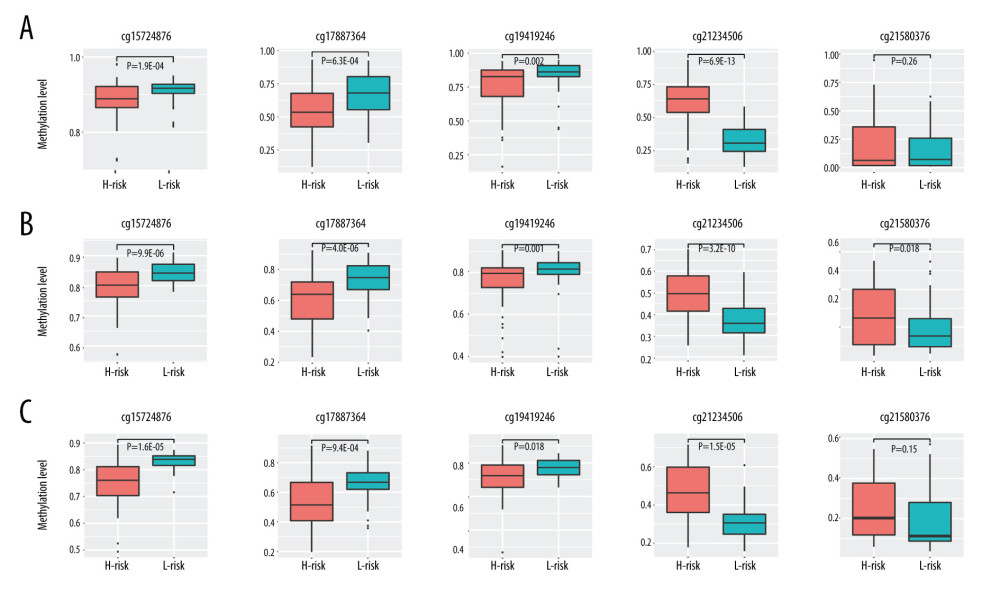 Figure 6. Methylation difference analysis of 5 differentially methylated sites within different risk groups in TCGA-TNBC (A), GSE141441 (B), and GSE78754 (C).
Figure 6. Methylation difference analysis of 5 differentially methylated sites within different risk groups in TCGA-TNBC (A), GSE141441 (B), and GSE78754 (C). 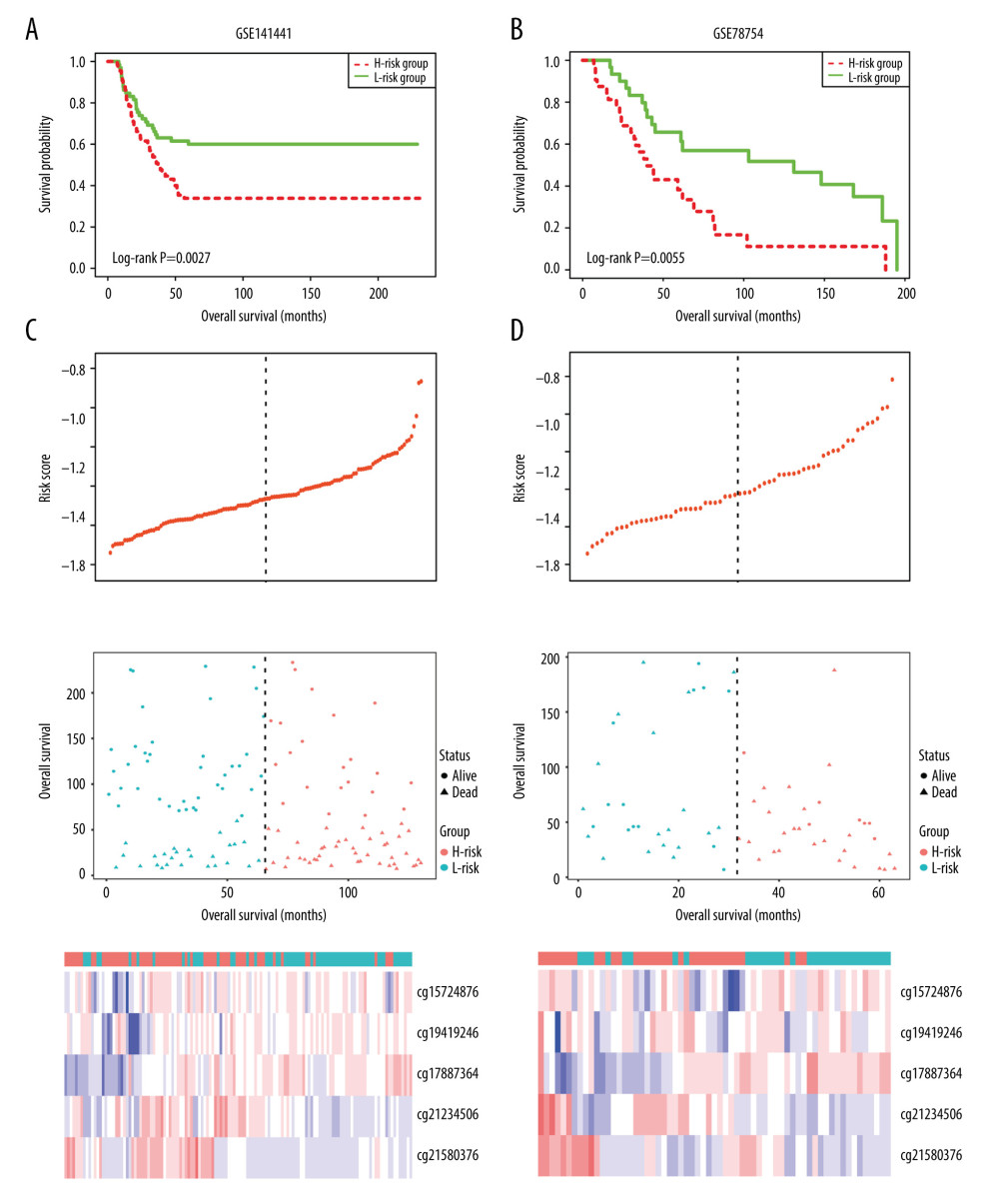 Figure 7. Kaplan-Meier survival curve in GSE141441 (A) and GSE78754 (B), and distribution of risk score, survival status, and methylation heatmap of 5 DMSs in GSE141441 (C) and GSE78754 (D).
Figure 7. Kaplan-Meier survival curve in GSE141441 (A) and GSE78754 (B), and distribution of risk score, survival status, and methylation heatmap of 5 DMSs in GSE141441 (C) and GSE78754 (D). Tables
Table 1. Cohorts analyzed in the present study.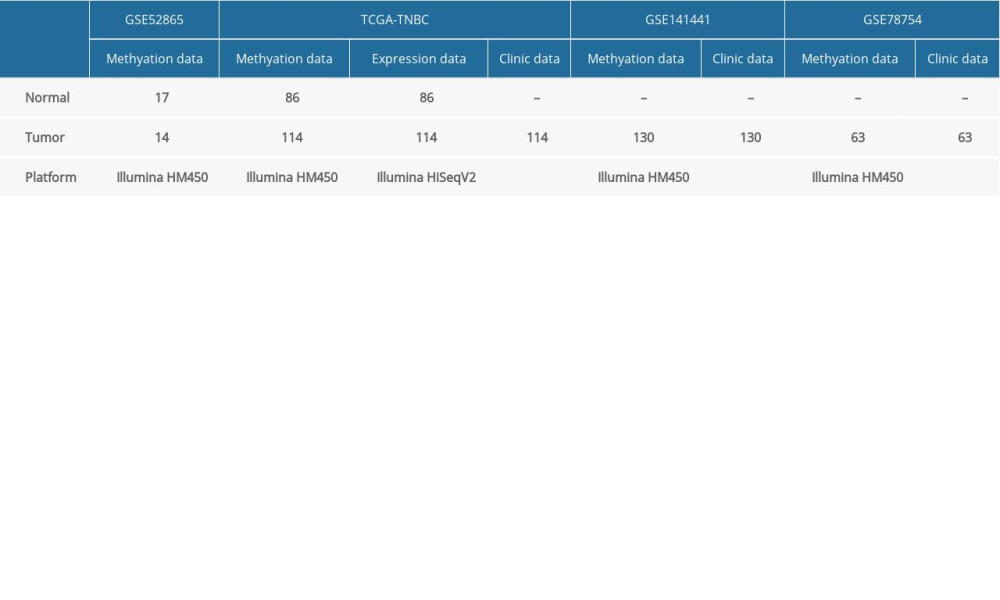 Table 2. Clinical characteristics of the patients in the training and validation cohorts.
Table 2. Clinical characteristics of the patients in the training and validation cohorts.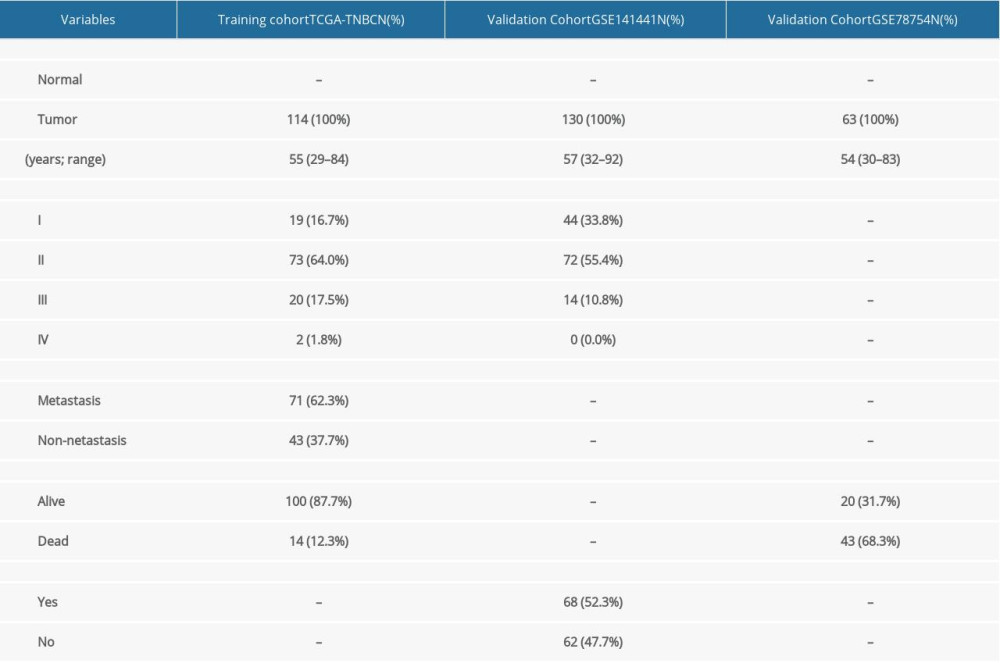 Table 3. General information of the 5 differentially methylated sites.
Table 3. General information of the 5 differentially methylated sites.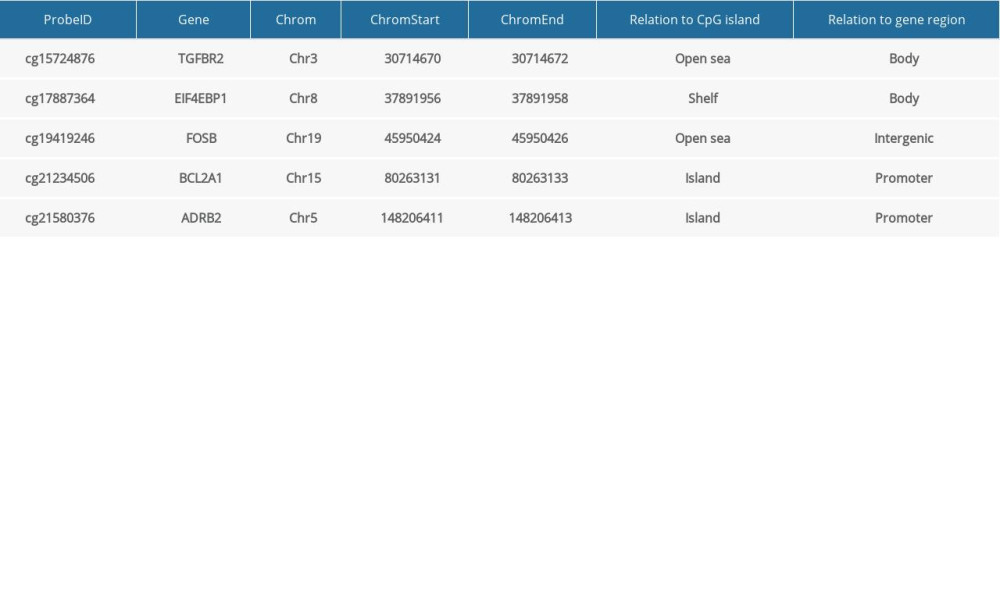 Table 4. Univariate and multivariate Cox regression analysis in the training and validation cohorts.
Table 4. Univariate and multivariate Cox regression analysis in the training and validation cohorts.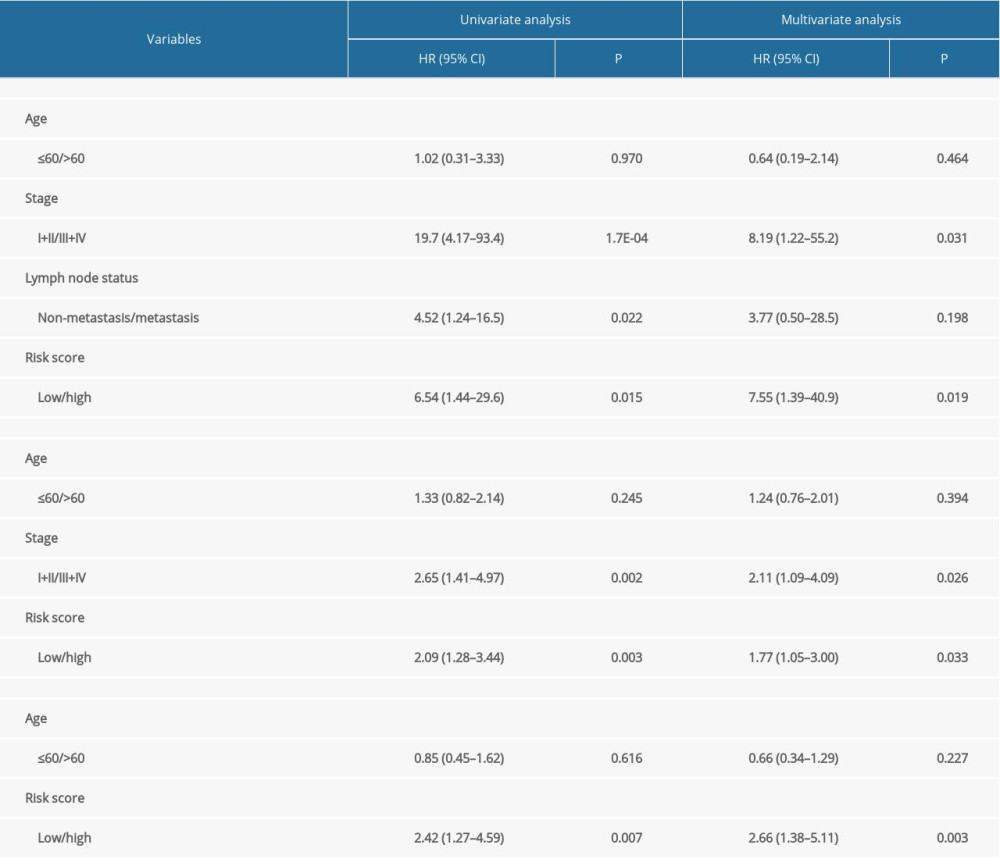 Supplementary Table 1. List of prognosis-related differentially methylated sites.
Supplementary Table 1. List of prognosis-related differentially methylated sites.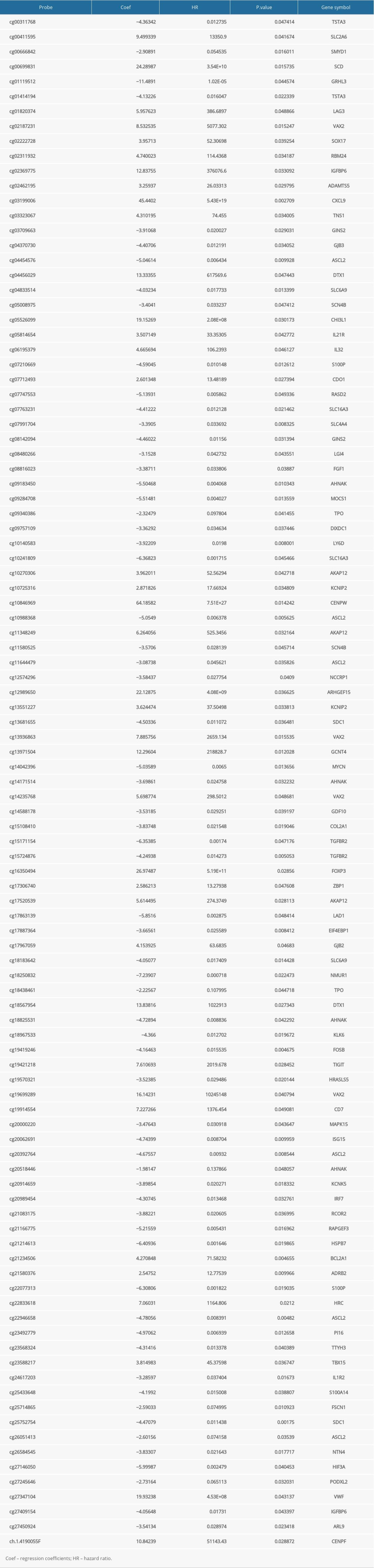
References
1. Bray F, Ferlay J, Soerjomataram I, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries: Cancer J Clin, 2018; 68(6); 394-424
2. Harbeck N, Gnant M, Breast cancer: Lancet, 2017; 389(10074); 1134-50
3. Dent R, Trudeau M, Pritchard KI, Triple-negative breast cancer: Clinical features and patterns of recurrence: Clin Cancer Res, 2007; 13(15 Pt 1); 4429-34
4. Qiu J, Xue X, Hu C, Comparison of clinicopathological features and prognosis in triple-negative and non-triple negative breast cancer: J Cancer, 2016; 7(2); 167-73
5. Rana AK, Ankri S, Reviving the RNA world: An insight into the appearance of RNA methyltransferases: Front Genet, 2016; 7; 99
6. Jaenisch R, Bird A, Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals: Nat Genet, 2003(33 Suppl); 245-54
7. Serman A, Vlahovic M, Serman L, Bulic-Jakus F, DNA methylation as a regulatory mechanism for gene expression in mammals: Coll Antropol, 2006; 30(3); 665-71
8. Weber M, Hellmann I, Stadler MB, Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome: Nat Genet, 2007; 39(4); 457-66
9. Stein R, Razin A, Cedar H, In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells: Proc Natl Acad Sci USA, 1982; 79(11); 3418-22
10. Hansen RS, Gartler SM, 5-Azacytidine-induced reactivation of the human X chromosome-linked PGK1 gene is associated with a large region of cytosine demethylation in the 5′ CpG island: Proc Natl Acad Sci USA, 1990; 87(11); 4174-78
11. Akhavan-Niaki H, Samadani AA, DNA methylation and cancer development: Molecular mechanism: Cell Biochem Biophys, 2013; 67(2); 501-13
12. Dehan P, Kustermans G, Guenin S, DNA methylation and cancer diagnosis: new methods and applications: Expert Rev Mol Diagn, 2009; 9(7); 651-57
13. Klutstein M, Nejman D, Greenfield R, Cedar H, DNA methylation in cancer and aging: Cancer Res, 2016; 76(12); 3446-50
14. Nakai K, Xia W, Liao HW, The role of PRMT1 in EGFR methylation and signaling in MDA-MB-468 triple-negative breast cancer cells: Breast Cancer, 2018; 25(1); 74-80
15. Xu J, Sun T, Guo X, Estrogen receptor-alpha promoter methylation is a biomarker for outcome prediction of cisplatin resistance in triple-negative breast cancer: Oncol Lett, 2018; 15(3); 2855-62
16. Prajzendanc K, Domagala P, Hybiak J, BRCA1 promoter methylation in peripheral blood is associated with the risk of triple-negative breast cancer: Int J Cancer, 2020; 146(5); 1293-98
17. Li C, Zheng Y, Pu K, A four-DNA methylation signature as a novel prognostic biomarker for survival of patients with gastric cancer: Cancer Cell Int, 2020; 20; 88
18. Tao C, Luo R, Song J, A seven-DNA methylation signature as a novel prognostic biomarker in breast cancer: J Cell Biochem, 2020; 121(3); 2385-93
19. Wang Y, Wang Y, Wang Y, Zhang Y, Identification of prognostic signature of non-small cell lung cancer based on TCGA methylation data: Sci Rep, 2020; 10(1); 8575
20. Peng Y, Shui L, Xie J, Liu S, Development and validation of a novel 15-CpG-based signature for predicting prognosis in triple-negative breast cancer: J Cell Mol Med Aug, 2020; 24(16); 9378-87
21. Carey LA, Dees EC, Sawyer L, The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes: Clin Cancer Res, 2007; 13(8); 2329-34
22. Liedtke C, Mazouni C, Hess KR, Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer: J Clin Oncol, 2008; 26(8); 1275-81
23. Braicu C, Chiorean R, Irimie A, Novel insight into triple-negative breast cancers, the emerging role of angiogenesis, and antiangiogenic therapy: Expert Rev Mol Med, 2016; 18; e18
24. Ribatti D, Nico B, Ruggieri S, Angiogenesis and antiangiogenesis in triple-negative breast cancer: Transl Oncol, 2016; 9(5); 453-57
25. Wang C, Li J, Ye S, Oestrogen inhibits VEGF expression and angiogenesis in triple-negative breast cancer by activating GPER-1: J Cancer, 2018; 9(20); 3802-11
26. Matsumoto H, Koo SL, Dent R, Role of inflammatory infiltrates in triple negative breast cancer: J Clin Pathol, 2015; 68(7); 506-10
27. Nakazawa Y, Taniyama Y, Sanada F, Periostin blockade overcomes chemoresistance via restricting the expansion of mesenchymal tumor subpopulations in breast cancer: Sci Rep, 2018; 8(1); 4013
28. Nekulova M, Holcakova J, Gu X, DeltaNp63alpha expression induces loss of cell adhesion in triple-negative breast cancer cells: BMC Cancer, 2016; 16(1); 782
29. Barbie TU, Alexe G, Aref AR, Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth: J Clin Invest, 2014; 124(12); 5411-23
30. Li M, Wang Y, Wei F, An X, Efficiency of cytokine-induced killer cells in combination with chemotherapy for triple-negative breast cancer: J Breast Cancer, 2018; 21(2); 150-57
31. Qiao Y, He H, Jonsson P, AP-1 is a key regulator of proinflammatory cytokine TNFalpha-mediated triple-negative breast cancer progression: J Biol Chem, 2016; 291(10); 5068-79
32. Khan MA, Jain VK, Rizwanullah M, PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges: Drug Discov Today, 2019; 24(11); 2181-91
33. Costa RLB, Han HS, Gradishar WJ, Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review: Breast Cancer Res Treat, 2018; 169(3); 397-406
34. Deng F, Weng Y, Li X, Overexpression of IL-8 promotes cell migration via PI3K-Akt signaling pathway and EMT in triple-negative breast cancer: Pathol Res Pract, 2020; 216(4); 152902
35. Sun J, Huang J, Lan J, Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer: Cancer Cell Int, 2019; 19; 264
36. Ueda A, Oikawa K, Fujita K, Therapeutic potential of PLK1 inhibition in triple-negative breast cancer: Lab Invest, 2019; 99(9); 1275-86
37. Zhang R, Li X, Liu Z, EZH2 inhibitors-mediated epigenetic reactivation of FOSB inhibits triple-negative breast cancer progress: Cancer Cell Int, 2020; 20; 175
38. Ma Y, He S, Gao A, Methylation silencing of TGF-beta receptor type II is involved in malignant transformation of esophageal squamous cell carcinoma: Clin Epigenetics, 2020; 12(1); 25
39. Xie F, Jin K, Shao L, FAF1 phosphorylation by AKT accumulates TGF-beta type II receptor and drives breast cancer metastasis: Nat Commun, 2017; 8; 15021
40. Coleman LJ, Peter MB, Teall TJ, Combined analysis of eIF4E and 4E-binding protein expression predicts breast cancer survival and estimates eIF4E activity: Br J Cancer, 2009; 100(9); 1393-99
41. Hiraki M, Maeda T, Mehrotra N, Targeting MUC1-C suppresses BCL2A1 in triple-negative breast cancer: Signal Transduct Target Ther, 2018; 3; 13
42. Kurozumi S, Kaira K, Matsumoto H, beta2-Adrenergic receptor expression is associated with biomarkers of tumor immunity and predicts poor prognosis in estrogen receptor-negative breast cancer: Breast Cancer Res Treat, 2019; 177(3); 603-10
43. Ting CH, Chen YC, Wu CJ, Targeting FOSB with a cationic antimicrobial peptide, TP4, for treatment of triple-negative breast cancer: Oncotarget, 2016; 7(26); 40329-47
44. Karlsson E, Perez-Tenorio G, Amin R, The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: a retrospective study including patients from the randomised Stockholm tamoxifen trials: Breast Cancer Res, 2013; 15(5); R96
Figures
Figure 1. Consistency of differentially methylated sites and identification of differentially expressed genes. (A) Overlapping differentially methylated sites between TCGA-TNBC and GSE52865. (B) Volcano plot of differentially expressed genes.Figure 2. Functional enrichment of the significantly enriched Gene Ontology terms (A) and Kyoto Encyclopedia of Genes and Genomes pathways (B) of differentially expressed genes containing differentially methylated sites.Figure 3. Identification of hub genes within protein-protein interaction (PPI) network. (A) PPI network. (B) Top 10 hub genes with high degrees of connectivity.Figure 4. Evaluating the prognostic power of the 5-DMSs signature in the TCGA-TNBC cohort. (A) Kaplan-Meier survival curve of the high- and low-risk groups. (B) Multivariate analysis of risk factors for TNBC. (C) Distribution of risk score, survival status, and methylation heatmap of the 5-DMSs.Figure 5. The stratification analysis for stage in TCGA-TNBC (A) and GSE141441 (B).Figure 6. Methylation difference analysis of 5 differentially methylated sites within different risk groups in TCGA-TNBC (A), GSE141441 (B), and GSE78754 (C).Figure 7. Kaplan-Meier survival curve in GSE141441 (A) and GSE78754 (B), and distribution of risk score, survival status, and methylation heatmap of 5 DMSs in GSE141441 (C) and GSE78754 (D). Tables
Table 1. Cohorts analyzed in the present study.Table 2. Clinical characteristics of the patients in the training and validation cohorts.Table 3. General information of the 5 differentially methylated sites.Table 4. Univariate and multivariate Cox regression analysis in the training and validation cohorts.Table 1. Cohorts analyzed in the present study.Table 2. Clinical characteristics of the patients in the training and validation cohorts.Table 3. General information of the 5 differentially methylated sites.Table 4. Univariate and multivariate Cox regression analysis in the training and validation cohorts.Supplementary Table 1. List of prognosis-related differentially methylated sites. In Press
15 Apr 2024 : Laboratory Research
The Role of Copper-Induced M2 Macrophage Polarization in Protecting Cartilage Matrix in OsteoarthritisMed Sci Monit In Press; DOI: 10.12659/MSM.943738
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952