20 March 2021: Database Analysis
Potential Target Genes in the Development of Atrial Fibrillation: A Comprehensive Bioinformatics Analysis
Liang Liu1CE, Yun Yu1BC, Long-long Hu1D, Quan-bin Dong1D, Feng Hu1D, Ling-juan Zhu1D, Qian Liang1D, Ling-ling Yu1B, Hui-hui Bao1A, Xiao-shu Cheng1A*DOI: 10.12659/MSM.928366
Med Sci Monit 2021; 27:e928366






Abstract
BACKGROUND: Atrial fibrillation (AF) is the most prevalent arrhythmia worldwide. Although it is not life-threatening, the accompanying rapid and irregular ventricular rate can lead to hemodynamic deterioration and obvious symptoms, especially the risk of cerebrovascular embolism. Our study aimed to identify novel and promising genes that could explain the underlying mechanism of AF development.
MATERIAL AND METHODS: Expression profiles GSE41177, GSE79768, and GSE14975 were acquired from the Gene Expression Omnibus Database. R software was used for identifying differentially expressed genes (DEGs), and Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analyses were subsequently performed. A protein–protein interaction network was constructed in Cytoscape software. Next, a least absolute shrinkage and selection operator (LASSO) model was constructed and receiver-operating characteristic curve analysis was conducted to assess the specificity and sensitivity of the key genes.
RESULTS: We obtained 204 DEGs from the datasets. The DEGs were mostly involved in immune response and cell communication. The primary pathways of the DEGs were related to the course or maintenance of autoimmune and chronic inflammatory diseases. The top 20 hub genes (high scores in cytoHubba) were selected in the PPI network. Finally, we identified 6 key genes (FCGR3B, CLEC10A, FPR2, IGSF6, S100A9, and S100A12) via the LASSO model.
CONCLUSIONS: We present 6 target genes that are potentially involved in the molecular mechanisms of AF development. In addition, these genes are likely to serve as potential therapeutic targets.
Keywords: Atrial Fibrillation, Biological Markers, Immunity, Active, Biomarkers, Tumor, Computational Biology, Databases, Genetic, Gene Expression Profiling, gene ontology, Gene Regulatory Networks, Protein Interaction Mapping, Protein Interaction Maps, Software
Background
Atrial fibrillation (AF) is the most prevalent (0.51%) and persistent heart rhythm disorder globally [1]. AF, increases the risk of blood clots causing thromboembolism, confers a nearly 5-fold increased risk of a debilitating stroke, and is a major risk factor for cardiovascular outcomes, including stroke [2,3]. Given its asymptomatic nature, many cases of AF go undetected until complications occur. Early detection of AF increases the chances of preventing stroke and other complications [4]. Therefore, it is imperative to explore the molecular mechanisms of AF pathogenesis as a means of improving the early diagnosis and treatment interventions of the disease.
Changes in genetic expression in AF are gradually gaining research attention [5,6]. Using the expression profiles of AF-associated target genes as a guide to examine the pathogenic sites of genetic variation and target gene regulation could be an effective approach [7,8]. Population studies show that a positive family history of AF increases the risk of developing AF among first-degree relatives by 30% [9,10]. The search for AF-associated genetic loci is propelled by the existing knowledge on the heritability of AF in the general population. Several genes, including some related to ion channel function, have been revealed to be associated with AF [11]. Ion channel-related genes associated with AF are primarily related to potassium channels. Previous studies have identified specific potassium channel genes or subunits that are associated AF. Monogenic AF pedigrees revealed that mutations leading to the gain or loss of function of
In the present study, based on a comprehensive bioinformatics analysis, we identified DEGs between sinus rhythm (SR) and AF samples, and further elucidated their potential molecular mechanisms and pathology in AF. This study adds several new candidate genes and related molecular pathways to those already associated with AF in previous studies. In future studies, these candidate genes and pathways could be investigated more closely to identify new and clear gene targets and potentially provide guidance for subsequent clinical studies.
Material and Methods
DATA INFORMATION AND PROCESSING:
The gene expression profiles of the GSE41177, GSE79768, and GSE14975 datasets were retrieved from the National Center for Biotechnology Information Gene Expression Omnibus database (NCBI GEO,
IDENTIFICATION OF DEGS:
Gene annotation was conducted before the analysis of the DEGs between AF and SR samples. The probe identification numbers of the merged data were matched with the gene symbols using the affy package (
GENE ONTOLOGY AND PATHWAY ENRICHMENT ANALYSIS:
Based on the analysis of DEGs, added potential functional annotations in Functional Enrichment analysis tool (Funrich) were performed. Gene Ontology (GO) term enrichment analysis, included biological process (BP), cellular component (CC), and molecular function (MF). In addition, the Database for Annotation, Visualization, and Integrated Discovery (DAVID, version 6.8,
ANALYSIS OF PROTEIN–PROTEIN INTERACTION NETWORKS OF DEGS:
The protein–protein interaction (PPI) networks of the DEGs were analyzed using the STRING online database (version 10.5;
CONSTRUCTION OF THE LASSO MODEL AND ROC CURVE ANALYSIS:
Based on the hub genes, we constructed a least absolute shrinkage and selection operator (LASSO) model because of its strong predictive value using the glmnet package (
Results
IDENTIFICATION OF DEGS IN AF:
We obtained the microarray datasets GSE41177, GSE79768, and GSE14975 from the GEO database. The datasets contained 51 AF and 23 SR samples. With the cutoff criteria set at adj. P<0.05 and |log2 FC| >1, we identified 204 DEGs (Figure 1). Compared with the control SR differential genes in the datasets, AF-related genes were mostly upregulated. The expression heatmaps of the DEGs are shown in Figure 1A, 1B. These genes were well clustered between the AF and control cases.
GO TERM ENRICHMENT ANALYSIS OF DEGS:
We identified the DEGs for the GO analysis using the Funrich software, and the DEGs were examined based on 3 categories: BP, CC, and MF. In the BP group, the DEGs were mainly enriched in immune response, cell communication, and signal transduction; in the CC group, the DEGs were primarily enriched in plasma membrane and exosomes; and in the MF group, the enrichment of the DEGs was mostly in the receptor activity, MHC class I receptor activity, and MHC class II receptor activity (Figure 2A–2C, Table 1).
SIGNALING PATHWAY ENRICHMENT ANALYSIS:
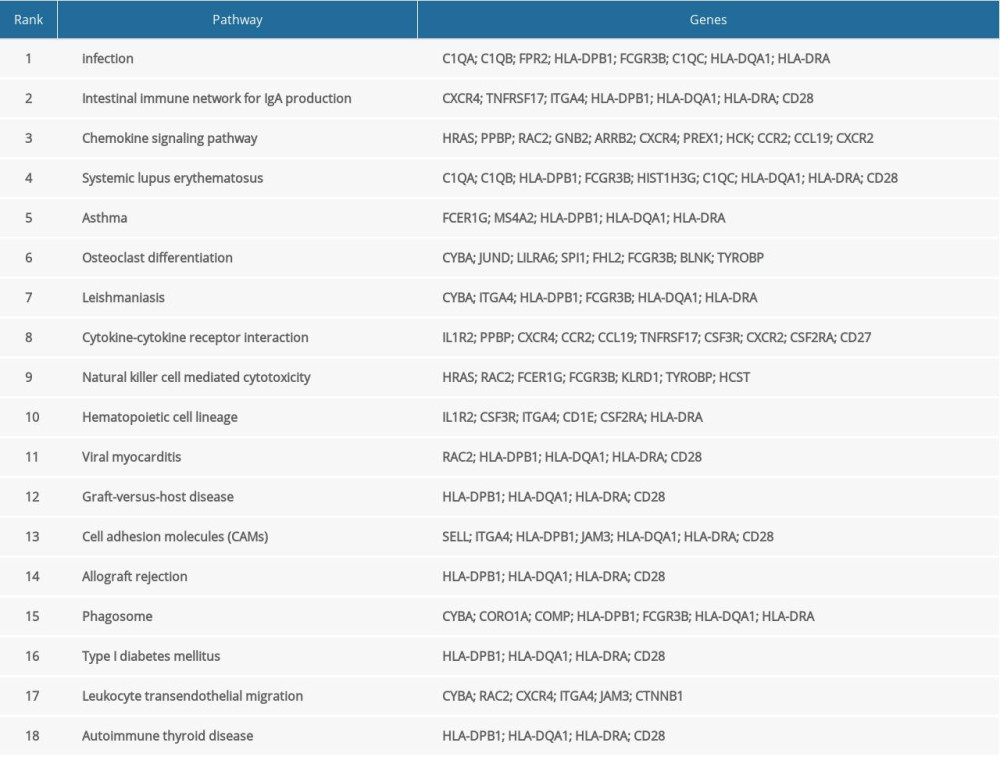
The KEGG pathway enrichment analysis was performed for the DEGs, and the results are shown in Figure 2D and Table 2. Enrichment of the DEGs was mainly in the Staphylococcus aureus infection, intestinal immune network for IgA production, systemic lupus erythematosus, asthma, and viral myocarditis.
PPI NETWORK AND HUB GENES IDENTIFICATION:
Using the STRING database and Cytoscape software, we constructed 4 PPI networks for the DEGs as shown in Figure 3A–3E. Furthermore, the top 20 hub genes of each dataset were identified using the 4 algorithms (Stress, Closeness, EcCentricity, and MCC) of the cytoHubba plugin. Among these hub genes, SPI1, HCK, TLR8, FPR2, RAC2, IGSF6, CSF3R, CXCR4, S100A8, S100A9, FCGR3B, TYROBP, S100A12, and CLEC10A exhibited the highest degree of association with AF and are listed in the Venn diagram (Figure 3F).
VALIDATION OF THE DIAGNOSTIC VALUE OF KEY GENES:
The construction of a LASSO model was based on the A gene expression profile of the 14 hub genes (Figure 4A). Based on the model, 6 genes were identified according to the regression coefficients that were not equal to zero. ROC curve analysis (Figure 4B, 4C) identified the AUC for FCGR3B, CLEC10A, FPR2, IGSF6, S100A9, and S100A12 in AF patients and normal controls; in the training set, the AUC values were 0.9981 (95% confidence interval [CI], 0.993–1), 0.9944 (95% CI, 0.9821–1), 0.987 (95% CI, 0.9655–1), 0.9481 (95% CI, 0.8566–1), 0.9852 (95% CI, 0.9616–1), and 0.9981 (95% CI, 0.993–1); in the test set, the AUC values were 0.8762 (95% CI, 0.7271–1), 0.9714 (95% CI, 0.9079–1), 0.981 (95% CI, 0.9358–1), 0.8857 (95% CI, 0.7092–1), 0.8952 (95% CI, 0.7517–1), and 0.9429 (95% CI, 0.825–1).
Discussion
The initiation and development of AF are regulated by the expression of many genes. Here, we systematically investigated gene expression profiles obtained from AF microarray studies. Gene expression data were downloaded from 3 GEO datasets in the GCBI. To better explore the DEGs, we identified the potential and remarkable DEGs, biological pathways, and processes based on comparisons between AF and normal SR samples.
GO analysis revealed that a considerable number of the co-expressed DEGs were primarily associated with immune response, cell communication, signal transduction, plasma membrane, exosomes, receptor activity, MHC class I receptor activity, and MHC class II receptor activity. Previous studies indicated that the immune response can be a complex and powerful factor in the pathophysiological process of AF and its concomitant complications [23,24]. The major immune cells found in the left atrial appendages of patients with AF were immunologically active monocytes and macrophages, suggesting a proinflammatory state characterized by increased infiltration of iNOS-positive, but Arg1-negative immune cells [25,26]. AF-induced cardiac injury processes of the myocardium initiate a cardiac immune response as shown by damage-associated molecular patterns (DAMPs), such as extracellular matrix and reactive oxygen species [27]. The pattern recognition receptor then initiates recognition of DAMPs, before the activation of innate and adaptive immune cells. This immune process forms a long-term feedback loop and promotes the formation of permanent forms of AF [28]. Inflammation and immune response are dependent on the onset and maintenance of AF, which is in turn dependent on inflammation and immune response. In addition, a clinical phenomenon that AF promotes inflammation supports a vicious cycle of “AF begets AF” [29]. The results of our pathway analysis showed that the DEGs identified were mainly associated with the course or maintenance of autoimmune diseases and chronic inflammatory diseases. Multiple systems of systemic lupus erythematosus (SLE), a chronic inflammatory autoimmune disease, may be an independent risk factor for AF since SLE has cardiac manifestations. Recent studies have shown that the disease status of SLE is independently associated with AF after adjusting for age, sex, race, and coronary artery disease [30]. Moreover, systemic inflammation and disease pathways of chronic inflammatory diseases are considered as pathogenic contributors to the initiation and development of AF [31,32]. Chemokines guide the migration and activation of systemic leukocytes and may influence AF development [33]. Some studies have suggested that the level of inflammatory cytokines/chemokines is related to the progression of AF to myocardial fibrosis [34]. The expression of cytokines in AF promotes the degree of the recruitment of immune cells [35]. As a classic way related in immune reaction, transforming growth factor (TGF)-β and interleukin (IL)-6 were expressed more frequently in the atria of macrophages after migration in AF [36].
Further analysis of the co-expression genes in the PPI network and LASSO model identified 6 key genes (
Although the potential role of
CLEC10A is one of the members of the c-type lectin domain family 12, which serves as a characteristic galactose lectin on macrophages and dendritic cells. CLEC10A can be activated through Toll-like receptor signaling and increase the secretion of various cytokines, including tumor necrosis factor-α, IL-8, and IL-10 [49]. In addition, lectin has been linked to the development of AF, and it can particularly predict thrombosis and left atrial appendage remodeling in patients with AF [50]. A synergistic interaction exists between lectin and TGF-β1, which can induce AF by activating the TGF-β1/Smad pathway in patients with AF [51]. Formyl peptide receptors (FPRs) belong to a G-protein-coupled chemokine receptor family and include 3 subtypes (FPR1, FPR2, and FPR3) that play important roles in host defense and inflammation [52]. FPR2 is involved in the initiation and resolution of inflammation. Similarly, the FPR2 signaling has important vascular effects through the inflammatory response [53,54].
Conclusions
In summary, we used bioinformatics analysis tools to delineate the pathomechanism of AF based on DEGs. Autoimmune and chronic inflammatory pathways, neutrophil chemotaxis, and immune response were confirmed to play roles in the development of AF. In addition, 6 genes, including
There are some limitations of our study. First, the clinical atrial tissue samples were not analyzed in the laboratory. Therefore, future studies on the mechanism of AF occurrence and development should incorporate such samples. In addition, given the current limited datasets, the sample sizes were relatively small and a multicenter study involving a larger sample size is needed. Further investigation is required to determine whether the 6 key genes (
Figures
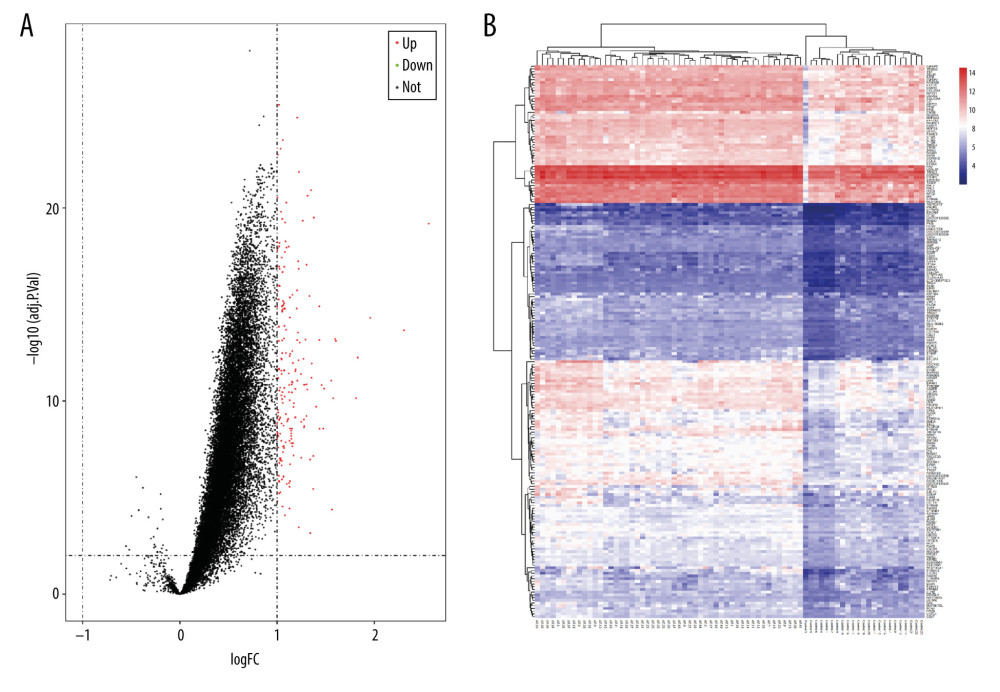 Figure 1. (A) Volcano plot of potential differentially expressed genes (DEGs). Red dots denote upregulated genes; green dots denote downregulated genes. (B) Heat maps for the DEGs between atrial fibrillation (AF) and sinus rhythm (SR).
Figure 1. (A) Volcano plot of potential differentially expressed genes (DEGs). Red dots denote upregulated genes; green dots denote downregulated genes. (B) Heat maps for the DEGs between atrial fibrillation (AF) and sinus rhythm (SR). 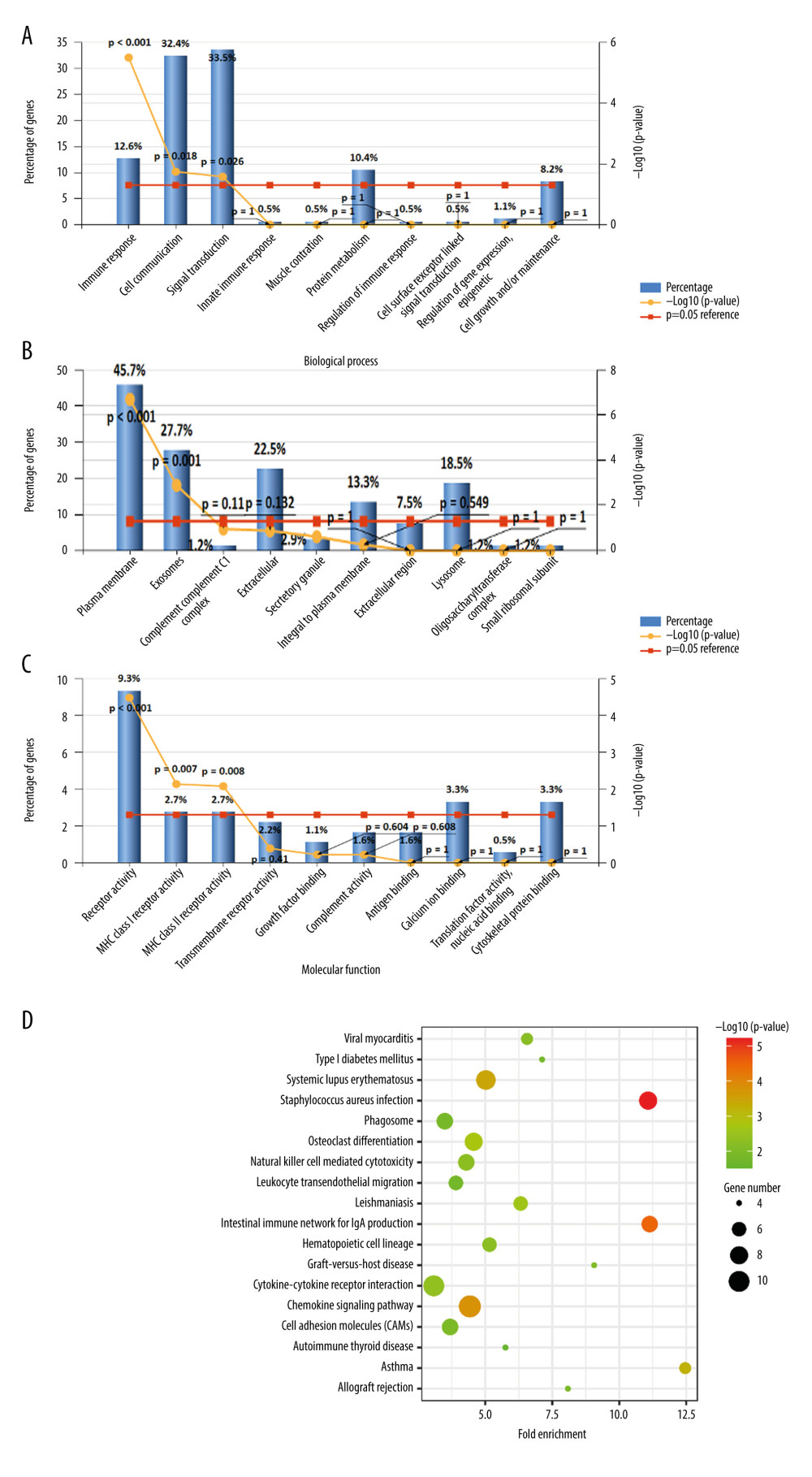 Figure 2. Biological functions based on Gene Ontology (GO) analysis of the differentially expressed genes (DEGs): (A) biological process (BP); (B) cellular component (CC), and (C) molecular function (MF). (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the DEGs.
Figure 2. Biological functions based on Gene Ontology (GO) analysis of the differentially expressed genes (DEGs): (A) biological process (BP); (B) cellular component (CC), and (C) molecular function (MF). (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the DEGs. 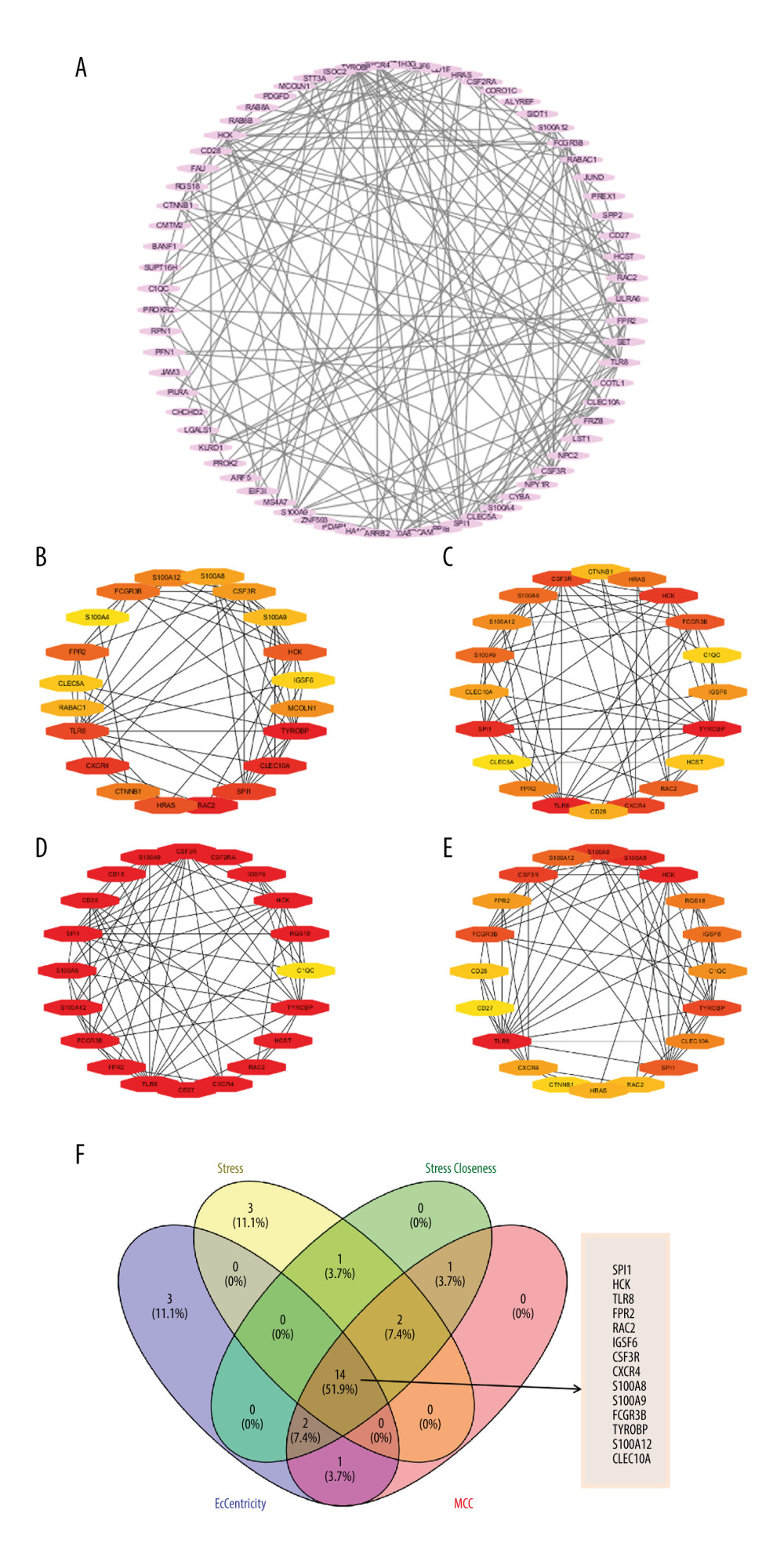 Figure 3. (A) Protein–protein interaction (PPI) networks of the differentially expressed genes (DEGs). The top 20 genes in cytoHubba plugins: (B) Stress, (C) Closeness, (D) EcCentricity, and (E) MCC. (F) Venn diagrams of the top 20 genes.
Figure 3. (A) Protein–protein interaction (PPI) networks of the differentially expressed genes (DEGs). The top 20 genes in cytoHubba plugins: (B) Stress, (C) Closeness, (D) EcCentricity, and (E) MCC. (F) Venn diagrams of the top 20 genes. 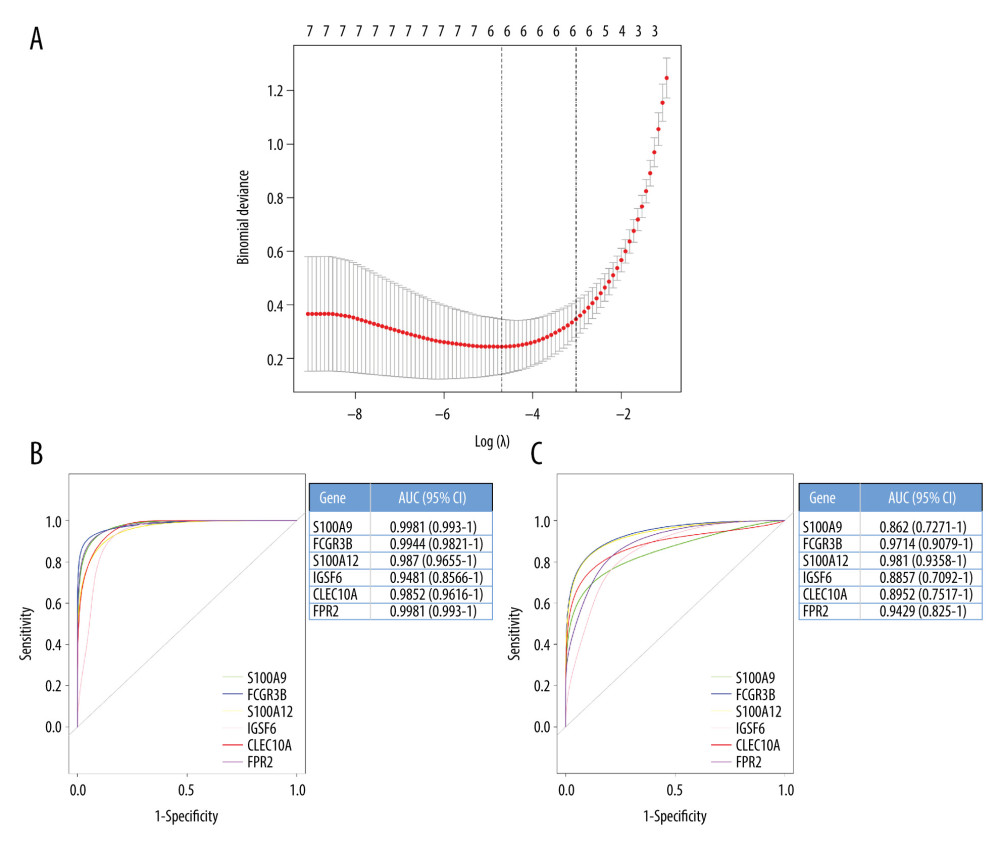 Figure 4. (A) Least absolute shrinkage and selection operator (LASSO) regression analysis of the hub genes. (B) Receiver-operating characteristic (ROC) curve of the hub genes in the training cohort. (C) ROC curve of the hub genes in the testing cohort.
Figure 4. (A) Least absolute shrinkage and selection operator (LASSO) regression analysis of the hub genes. (B) Receiver-operating characteristic (ROC) curve of the hub genes in the training cohort. (C) ROC curve of the hub genes in the testing cohort. 
References
1. Lippi G, Sanchis-Gomar F, Cervellin G, Global epidemiology of atrial fibrillation: An increasing epidemic and public health challenge: Int J Stroke, 2020 [Online ahead of print]
2. Soo Y, Chan N, Leung KT, Age-specific trends of atrial fibrillation-related ischaemic stroke and transient ischaemic attack, anticoagulant use and risk factor profile in Chinese population: A 15-year study: J Neurol Neurosurg Psychiatry, 2017; 88(9); 744-48
3. Wang X, Fu Q, Song F, Prevalence of atrial fibrillation in different socioeconomic regions of China and its association with stroke: Results from a national stroke screening survey: Int J Cardiol, 2018; 271; 92-97
4. Esato M, Chun YH, An Y, Clinical impact of asymptomatic presentation status in patients with paroxysmal and sustained atrial fibrillation: The Fushimi AF Registry: Chest, 2017; 152(6); 1266-75
5. Hucker WJ, Hanley A, Ellinor PT, Improving atrial fibrillation therapy: Is there a gene for that?: J Am Coll Cardiol, 2017; 69(16); 2088-95
6. Roberts JD, Gollob MH, A contemporary review on the genetic basis of atrial fibrillation: Methodist Debakey Cardiovasc J, 2014; 10(1); 18-24
7. van Ouwerkerk AF, Bosada FM, van Duijvenboden K, Identification of atrial fibrillation associated genes and functional non-coding variants: Nat Commun, 2019; 10(1); 4755
8. Zhang M, Hill MC, Kadow ZA, Long-range Pitx2c enhancer-promoter interactions prevent predisposition to atrial fibrillation: Proc Natl Acad Sci USA, 2019; 116(45); 22692-98
9. McAlister FA, Yan L, Roos LL, Lix LM, Parental atrial fibrillation and stroke or atrial fibrillation in young adults: Stroke, 2019; 50(9); 2322-28
10. Fox CS, Parise H, D’Agostino RS, Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring: JAMA, 2004; 291(23); 2851-55
11. Campbell HM, Wehrens X, Genetics of atrial fibrillation: An update: Curr Opin Cardiol, 2018; 33(3); 304-10
12. Chen YH, Xu SJ, Bendahhou S, KCNQ1 gain-of-function mutation in familial atrial fibrillation: Science, 2003; 299(5604); 251-54
13. Voudris KV, Apostolakis S, Karyofillis P, Genetic diversity of the KCNE1 gene and susceptibility to postoperative atrial fibrillation: Am Heart J, 2014; 167(2); 274-80.e1
14. Olesen MS, Bentzen BH, Nielsen JB, Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation: BMC Med Genet, 2012; 13; 24
15. Yamada N, Asano Y, Fujita M, Mutant KCNJ3 and KCNJ5 potassium channels as novel molecular targets in bradyarrhythmias and atrial fibrillation: Circulation, 2019; 139(18); 2157-69
16. Chang SN, Tsai CT, Wu CK, A functional variant in the promoter region regulates the C-reactive protein gene and is a potential candidate for increased risk of atrial fibrillation: J Intern Med, 2012; 272(3); 305-15
17. Chang SN, Lai LP, Chiang FT, C-reactive protein gene polymorphism predicts the risk of thromboembolic stroke in patients with atrial fibrillation: A more than 10-year prospective follow-up study: J Thromb Haemost, 2017; 15(8); 1541-46
18. Beyer EC, Berthoud VM, Gap junction gene and protein families: Connexins, innexins, and pannexins: Biochim Biophys Acta Biomembr, 2018; 1860(1); 5-8
19. Bikou O, Thomas D, Trappe K, Connexin 43 gene therapy prevents persistent atrial fibrillation in a porcine model: Cardiovasc Res, 2011; 92(2); 218-25
20. Mao L, Huang W, Zou P, The unrecognized role of tumor suppressor genes in atrial fibrillation: Gene, 2018; 642; 26-31
21. Zhou R, Liu Y, Huang W, Dang X, Potential functions of esophageal cancer-related gene-4 in the cardiovascular system: Front Med, 2019; 13(6); 639-45
22. Huang L, Yu H, Fan X, A potential role of esophageal cancer related gene-4 for atrial fibrillation: Sci Rep, 2017; 7(1); 2717
23. Liu Y, Shi Q, Ma Y, Liu Q, The role of immune cells in atrial fibrillation: J Mol Cell Cardiol, 2018; 123; 198-208
24. Hu YF, Chen YJ, Lin YJ, Chen SA, Inflammation and the pathogenesis of atrial fibrillation: Nat Rev Cardiol, 2015; 12(4); 230-43
25. Yamashita T, Sekiguchi A, Iwasaki YK, Recruitment of immune cells across atrial endocardium in human atrial fibrillation: Circ J, 2010; 74(2); 262-70
26. Sun Z, Zhou D, Xie X, Cross-talk between macrophages and atrial myocytes in atrial fibrillation: Basic Res Cardiol, 2016; 111(6); 63
27. Tousoulis D, Zisimos K, Antoniades C, Oxidative stress and inflammatory process in patients with atrial fibrillation: The role of left atrium distension: Int J Cardiol, 2009; 136(3); 258-62
28. Boos CJ, Infection and atrial fibrillation: Inflammation begets AF: Eur Heart J, 2020; 41(10); 1120-22
29. Lo LW, Chang HY, Scherlag BJ, Temporary suppression of cardiac ganglionated plexi leads to long-term suppression of atrial fibrillation: Evidence of early autonomic intervention to break the vicious cycle of “AF begets AF”: J Am Heart Assoc, 2016; 5(7); e003309
30. Barnado A, Carroll RJ, Casey C, Phenome-wide association studies uncover a novel association of increased atrial fibrillation in male patients with systemic lupus erythematosus: Arthritis Care Res (Hoboken), 2018; 70(11); 1630-36
31. Guo Y, Lip GY, Apostolakis S, Inflammation in atrial fibrillation: J Am Coll Cardiol, 2012; 60(22); 2263-70
32. Aviles RJ, Martin DO, Apperson-Hansen C, Inflammation as a risk factor for atrial fibrillation: Circulation, 2003; 108(24); 3006-10
33. Huang J, Wu N, Xiang Y, Prognostic value of chemokines in patients with newly diagnosed atrial fibrillation: Int J Cardiol, 2020; 320; 83-89
34. Abe I, Teshima Y, Kondo H, Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation: Heart Rhythm, 2018; 15(11); 1717-27
35. Aulin J, Siegbahn A, Hijazi Z, Interleukin-6 and C-reactive protein and risk for death and cardiovascular events in patients with atrial fibrillation: Am Heart J, 2015; 170(6); 1151-60
36. Yamashita T, Sekiguchi A, Iwasaki YK, Recruitment of immune cells across atrial endocardium in human atrial fibrillation: Circ J, 2010; 74(2); 262-70
37. Nimmerjahn F, Ravetch JV, Fcgamma receptors as regulators of immune responses: Nat Rev Immunol, 2008; 8(1); 34-47
38. Weber F, Breustedt D, Schlicht S, First infusion reactions are mediated by FcγRIIIb and neutrophils: Pharm Res, 2018; 35(9); 169
39. Ishii Y, Schuessler RB, Gaynor SL, Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation: Circulation, 2005; 111(22); 2881-88
40. Schuessler RB, Ishii Y, Khagi Y, The effects of inflammation on heart rate and rhythm in a canine model of cardiac surgery: Heart Rhythm, 2012; 9(3); 432-39
41. Treffers LW, van Houdt M, Bruggeman CW, FcγRIIIb restricts antibody-dependent destruction of cancer cells by human neutrophils: Front Immunol, 2018; 9; 3124
42. Bournazos S, Bournazou I, Murchison JT, Copy number variation of FCGR3B is associated with susceptibility to idiopathic pulmonary fibrosis: Respiration, 2011; 81(2); 142-49
43. Coxon A, Cullere X, Knight S, Fc gamma RIII mediates neutrophil recruitment to immune complexes. A mechanism for neutrophil accumulation in immune-mediated inflammation: Immunity, 2001; 14(6); 693-704
44. McKinney C, Broen JC, Vonk MC, Evidence that deletion at FCGR3B is a risk factor for systemic sclerosis: Genes Immun, 2012; 13(6); 458-60
45. Fanciulli M, Norsworthy PJ, Petretto E, FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity: Nat Genet, 2007; 39(6); 721-23
46. Lee YH, Bae SC, Seo YH, Association between FCGR3B copy number variations and susceptibility to autoimmune diseases: A meta-analysis: Inflamm Res, 2015; 64(12); 983-91
47. Zaghlool SB, Kuhnel B, Elhadad MA, Epigenetics meets proteomics in an epigenome-wide association study with circulating blood plasma protein traits: Nat Commun, 2020; 11(1); 15
48. Suzuki A, Fukuzawa K, Yamashita T, Circulating intermediate CD14++CD16+monocytes are increased in patients with atrial fibrillation and reflect the functional remodelling of the left atrium: Europace, 2017; 19(1); 40-47
49. Heger L, Balk S, Luhr JJ, CLEC10A is a specific marker for human CD1c(+) dendritic cells and enhances their Toll-like receptor 7/8-induced cytokine secretion: Front Immunol, 2018; 9; 744
50. Tang Z, Zeng L, Lin Y, Circulating galectin-3 is associated with left atrial appendage remodelling and thrombus formation in patients with atrial fibrillation: Heart Lung Circ, 2019; 28(6); 923-31
51. Xiao M, Zhang M, Bie M, Galectin-3 induces atrial fibrosis by activating the TGF-beta1/Smad pathway in patients with atrial fibrillation: Cardiology, 2020; 145(7); 446-55
52. Senchenkova EY, Ansari J, Becker F, Novel role for the AnxA1-Fpr2/ALX signaling axis as a key regulator of platelet function to promote resolution of inflammation: Circulation, 2019; 140(4); 319-35
53. Petri MH, Laguna-Fernández A, Gonzalez-Diez M, The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability: Cardiovasc Res, 2015; 105(1); 65-74
54. Kain V, Jadapalli JK, Tourki B, Halade GV, Inhibition of FPR2 impaired leukocytes recruitment and elicited non-resolving inflammation in acute heart failure: Pharmacol Res, 2019; 146; 104295
Figures
Figure 1. (A) Volcano plot of potential differentially expressed genes (DEGs). Red dots denote upregulated genes; green dots denote downregulated genes. (B) Heat maps for the DEGs between atrial fibrillation (AF) and sinus rhythm (SR).Figure 2. Biological functions based on Gene Ontology (GO) analysis of the differentially expressed genes (DEGs): (A) biological process (BP); (B) cellular component (CC), and (C) molecular function (MF). (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the DEGs.Figure 3. (A) Protein–protein interaction (PPI) networks of the differentially expressed genes (DEGs). The top 20 genes in cytoHubba plugins: (B) Stress, (C) Closeness, (D) EcCentricity, and (E) MCC. (F) Venn diagrams of the top 20 genes.Figure 4. (A) Least absolute shrinkage and selection operator (LASSO) regression analysis of the hub genes. (B) Receiver-operating characteristic (ROC) curve of the hub genes in the training cohort. (C) ROC curve of the hub genes in the testing cohort. In Press
05 Mar 2024 : Clinical Research
Role of Critical Shoulder Angle in Degenerative Type Rotator Cuff Tears: A Turkish Cohort StudyMed Sci Monit In Press; DOI: 10.12659/MSM.943703
06 Mar 2024 : Clinical Research
Comparison of Outcomes between Single-Level and Double-Level Corpectomy in Thoracolumbar Reconstruction: A ...Med Sci Monit In Press; DOI: 10.12659/MSM.943797
21 Mar 2024 : Meta-Analysis
Economic Evaluation of COVID-19 Screening Tests and Surveillance Strategies in Low-Income, Middle-Income, a...Med Sci Monit In Press; DOI: 10.12659/MSM.943863
10 Apr 2024 : Clinical Research
Predicting Acute Cardiovascular Complications in COVID-19: Insights from a Specialized Cardiac Referral Dep...Med Sci Monit In Press; DOI: 10.12659/MSM.942612
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952