23 June 2020: Database Analysis
Bioinformatics-Based Study to Investigate Potential Differentially Expressed Genes and miRNAs in Pediatric Sepsis
Kexin Xie1BE, Shan Kong1CE, Fuxing Li1DF, Yulin Zhang1CD, Jing Wang2DF, Weidong Zhao1AG*DOI: 10.12659/MSM.923881
Med Sci Monit 2020; 26:e923881






Abstract
BACKGROUND: Sepsis is an extremely common health issue with a considerable mortality rate in children. Our understanding about the pathogenic mechanisms of sepsis is limited. The aim of this study was to identify the differential expression genes (DEGs) in pediatric sepsis through comprehensive analysis, and to provide specific insights for the clinical sepsis therapies in children.
MATERIAL AND METHODS: Three pediatric gene expression profiles (GSE25504, GSE26378, GSE26440) were downloaded from the Gene Expression Omnibus (GEO) database. The difference expression genes (DEGs) between pediatric sepsis and normal control group were screened with the GEO2R online tool. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the DEGs were performed. Cytoscape with CytoHubba were used to identify the hub genes. Finally, NetworkAnalyst was used to construct the targeted microRNAs (miRNAs) of the hub genes.
RESULTS: Totally, 160 overlapping upward genes and 61 downward genes were identified. In addition, 5 KEGG pathways, including hematopoietic cell lineage, Staphylococcus aureus infection, starch and sucrose metabolism, osteoclast differentiation, and tumor necrosis factor (TNF) signaling pathway, were significantly enriched using a database for labeling, visualization, and synthetic discovery. In combination with the results of the protein–protein interaction (PPI) network and CytoHubba, 9 hub genes including ITGAM, TLR8, IL1β, MMP9, MPO, FPR2, ELANE, SPI1, and C3AR1 were selected. Combined with DEG-miRNAs visualization, 5 miRNAs, including has-miR-204-5p, has-miR-211-5p, has-miR-590-5p, and has-miR-21-5p, were predicted as possibly the key miRNAs.
CONCLUSIONS: Our findings will contribute to identification of potential biomarkers and novel strategies for pediatric sepsis treatment.
Keywords: Computational Biology, Hu Paraneoplastic Encephalomyelitis Antigens, Sepsis, Child, Child, Preschool, Databases, Genetic, Gene Expression Profiling, gene ontology, Gene Regulatory Networks, Protein Interaction Maps
Background
Sepsis in children is regarded as the leading global cause of high mortality [1]. Every year, an estimated 1.2 million children are diagnosed with sepsis, with the mortality rate ranging from 4% to 50% [2–4]. Recently, expert consensus guidelines on the treatment of sepsis in children were proposed to assist clinicians in assessing pediatric sepsis and septic shock [5]. However, in view of the non-specific signs and symptoms of sepsis, the diagnosis and treatments of sepsis remain immensely complex.
Blood-based non-invasive biomarkers can be the key to personalized medicine in sepsis whereby patients receive specific treatments due to their identifiable molecular signatures [6]. Diverse biomarkers have been assessed in the diagnosis of pediatric sepsis [7–9]. However, none of these biomarkers has excellent specificity and/or sensitivity in the clinical practice. Procalcitonin (PCT) and C-reactive protein (CRP) have been treated as biomarkers for diagnosing sepsis in children because of their relatively better specificity [10]. Nevertheless, no gold standard biomarker currently exists for utility in routine medical settings. Therefore, identification of new biomarkers is urgently needed.
High-throughput technologies have been applied for the potential biomarker discoveries [11]. Clinical bioinformatics as an emerging strategy is considered one of the promising and pivotal approaches to overcome clinical challenges in early diagnosis, competent therapies, and predictive prognostication of cancer patients [12]. Such strategies have been used extensively for investigations in liver cancer, gastric cancer, and other types of cancer [13–15]. Also, an increasing number of novel biomarkers in some non-tumor diseases have been identified via bioinformatics analysis [16–18].
With this in mind, we made a preliminary attempt to elucidate biomarkers and the molecular mechanisms underlying sepsis. In this investigation, we downloaded 3 original datasets containing messenger RNA (mRNA) expression profiles of pediatric sepsis from the Gene Expression Omnibus (GEO) datasets for analysis. Then we identified differential expression genes (DEGs) between pediatric sepsis samples and normal samples. Subsequently, we built a protein–protein interaction (PPI) network and performed functions of genes and pathway enrichment analysis of the DEGs. Finally, potential microRNAs (miRNAs) associated with DEGs were predicted by constructing a miRNA-gene regulatory network through Cytoscape software.
Material and Methods
MICROARRAY DATA PROCESSING:
Three microarray datasets (GSE25504, GSE26378, GSE26440) of pediatric sepsis and control samples were collected from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) after a careful review. All data were freely downloaded via the internet. Details regarding these 3 gene expression profiles used for this analysis are presented in Table 1. Among them, GSE25504 was based on Illumina HumanHT-12 V3.0 expression beadchip. GSE26378 and GSE26440 were based on Agilent GPL570 platform ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array).
DIFFERENTIALLY EXPRESSED GENES (DEGS) SELECTION:
GEO2R (
FUNCTIONAL AND KEGG ENRICHMENT ANALYSIS OF DEGS:
Functional enrichment of DEGs was analyzed in 3 categories of the Gene Ontology (GO) domains: biological process (BP), cellular component (CC), and molecular function (MF). The KEGG database contains pathway datasets involving biological functions, diseases, chemicals, and drugs. In this investigation, significantly upregulated and downregulated DEGs combined sepsis microarray data were analyzed using an online software (Enrichr,
PROTEIN–PROTEIN INTERACTION (PPI) ESTABLISHMENT AND IDENTIFICATION OF HUB GENES:
An online tool (Search Tools for the Retrieval of Interacting Genes, STRING,
MIRNAS ASSOCIATED WITH HUB GENES:
The top 9 hub genes were mapped to corresponding miRNAs using NetworkAnalyst 3.0 (
Results
SEPSIS DEGS IDENTIFICATION:
Three datasets gene expression profiles (GSE25504, GSE26378, and GSE26440) were retrieved from those present in the GEO database including both healthy and sepsis samples. Among them, GSE25504 included 35 healthy samples and 28 sepsis samples, GSE26378 included 21 healthy samples and 82 sepsis samples, and GSE26440 included 32 healthy samples and 78 sepsis samples. According to the criteria of P<0.05 and |logFC| ≥1.0, 600 DEGs in total were obtained from GSE25504, containing 403 upregulated genes and 197 downregulated genes. The 1700 DEGs were obtained when the gene chip GSE26378 was filtered. Among them, 1065 genes were upregulated, and 635 genes were downregulated. In GSE26440 dataset, 1883 DEGs were obtained, 906 of which were upregulated and 977 of which were downregulated. All DEGs were compared between normal controls and sepsis samples. The gene expression profile of each 3 of DEGs containing 2 sets of sample data is shown in Figure 1.
These genes were further filtered and the Venn diagrams representing these gene were plotted. As presented in Figure 2, 221 DEGs were found to be remarkably differentially expressed among the 3 groups, with 160 upregulated genes and 61 downregulated genes (Table 2).
FUNCTIONAL CLASSIFICATION AND PATHWAY ENRICHMENT OF DEGS:
The major GO functional terms of the DEGs, including biological process (BP), molecular function (MF), cellular component (CC) ontologies, are illustrated as Figure 3. The top 5 significant GO-BP terms for DEGs were principally associated with neutrophil degranulation, neutrophil activation involved in immune response, neutrophil mediated immunity, membrane raft assembly, and complement receptor mediated signaling pathway (Figure 3A, Supplementary Table 1). The result showed that DEGs were primarily enriched in GO-MF, including the activities of complement receptor, phosphofructokinase activity, interleukin-1 receptor binding, C-C chemokine binding, and UDP-glucosyltransferase activity (Figure 3B, Supplementary Table 2). The analysis of GO-CC indicated that DEGs significantly enriched in specific granule, tertiary granule, T cell receptor complex, tertiary granule membrane, and specific granule lumen (Figure 3C, Supplementary Table 3). These results suggest that leukocyte degranulation, leukocyte mediated immunity, and leukocyte activation involved in immune response are the most significantly enriched functional terms for GO-BP, GO-MF, and GO-CC. The top 5 significant KEGG pathways of DEGs were enriched in human hematopoietic cell lineage, Staphylococcus aureus infection, starch and sucrose metabolism, osteoclast differentiation, and tumor necrosis factor (TNF) signaling pathway (Figure 3D, Supplementary Table 4).
PPI NETWORK CONSTRUCTION AND HUB GENES IDENTIFICATION:
On the basis of STRING database, the PPI analysis of DEGs were performed and visualized by Cytoscape as presented in Figure 4. The top 9 genes, including ITGAM, TLR8, IL1B, MMP9, MPO, FPR2, ELANE, SPI1, and C3AR1, were taken to as potential hub genes based on the node degree score generated via Cytoscape. These 9 hub genes are presented in Figure 5. The result shows that the integrin alpha m (ITGAM, degree 66) is the most significant gene, followed by Toll-like receptors (TLR8, degree 43), interleukin 1 beta (IL1β, degree 40), matrix metalloproteinases-9 (MMP9, degree 38), bone marrow peroxidase (MPO, degree 38), formyl peptide receptor 2 (FPR2, degree 37), neutrophil elastic enzyme (ELANE, degree 35), transcription factors pu.1 (SPI1, degree 35) 1, and complement component 3a receptor 1 (C3AR1, degree 34).
INTEGRATED MIRNA/GENE REGULATORY NETWORKS:
MiRNAs are known to regulate targeted genes transcription inhibition or abrogate protein translation. The significantly differential miRNAs and genes regulatory networks was constructed using Cytoscape software. The target miRNAs were predicted based on NetworkAnalyst databases. The top 9 DEGs and their corresponding regulatory miRNAs molecules are shown in Figure 6. For instance, among 9 DEGs, ITGAM, IL1B, and MMP9 could be predicted as common targets of 2 miRNAs (has-miR-204-5p and has-miR-211-5p). Two miRNAs (has-miR-590-5p and has-miR-21-5p) could be bind to 3 target genes (ITGAM, IL1B, and C3AR1). However, these findings need further validation.
Discussion
The pathogenesis of sepsis is multifactorial, with environmental and genetic factors interacting to produce some pathological features. In current investigation, 221 DEGs in total were identified, consisting of 160 upregulated genes and 61 downregulated genes. The results of GO functional classification indicated that the DEGs were mainly enriched in leukocyte degranulation, leukocyte mediated immunity, and leukocyte activation involved in immune response. In the PPI network of DEGs, 9 (ITGAM, TLR8, IL1β, MMP9, MPO, FPR2, ELANE, SPI1, and C3AR1) out of 221 genes had high degrees. These 9 genes were all upregulated in pediatric patients with sepsis. Most of these 9 hub genes have been shown to have key effects on leukocyte infiltration, and activation.
ITGAM (namely CD11b) regulates activation, adhesion, and migration of leucocytes from blood to the sites of injury [19,20]. Previous data by others suggested that increased expression of ITGAM on polymorphonuclear neutrophils (PMN) was correlated with the reduced survival of sepsis [21]. Blocking of ITGAM has been shown to significantly inhibit LPS-induced endotoxin shock and microbial sepsis [22].
TLR8, a member of Toll-like receptors (TLRs) family, is well known for its capacity to sense single stranded RNA (ssRNA) from viruses such as hepatitis B virus (HBV), hepatitis C virus (HCV), human immunodeficiency virus (HIV), and influenza virus. More recently, some investigations have confirmed that TLR8-mediated RNA recognition plays a vital role in sensing of
It has been established that IL1β has a powerful proinflammatory effect on host defense against some microbial pathogens [26]. It has been known that the IL-1 pathway regulates inflammation, angiogenesis, hematopoiesis, and cognition [27]. A clinal trial done in 2016 showed that the use of IL1 receptor inhibition was related to significant improvement of the 28-day survival of patients with sepsis [28]. Thus, IL1β may also contribute to the sepsis development.
MMP9 plays a vital role in encouraging inflammation and inhibiting platelet aggregation [29]. Previous studies have shown that the MMP9 levels in septic patients were significantly higher than that in healthy controls [30–32]. In further study, MMP9 expression levels was evaluated as prognostic biomarkers of sepsis, further indicating the potential for MMP9 in the pathophysiology of sepsis. MMP9 gene level disorders may participate in the pathophysiological process of sepsis and may act as a new molecular target for pediatric patients with sepsis diagnosis and prognosis.
Myeloperoxidase (MPO) is a sign of neutrophils infiltration and a sign of oxidative stress [33]. During sepsis, the production of MPO outpaces the body’s antioxidant defenses. This imbalance can lead to direct mitochondrial damages, which has been reported to result in sepsis-mediated organ failure [34,35]. Furthermore, other observations suggest that decreasing oxidative stress can serve as a safeguard strategy for mitochondria during sepsis [36,37]. MPO may therefore be a potentially new treatment target for pediatric sepsis.
Lipoxin A4 (LXA4), a kind of eicosanoid, is generated from arachidonic acid by sequential action of lipoxygenases [38]. Accumulating evidence have shown that activation of LXA4 receptor (ALX)/formyl peptide receptor 2 (FPR2) is essential in experimental sepsis models [39,40]. Additionally, ALX/FPR2 deficiency in mice abrogates bacterial clearance and provokes the host response in polymicrobial sepsis [41], which implies that ALX/FPR2 regulates anti-inflammatory reactions and may be a potential treatment option for pediatric sepsis.
Elastase neutrophil expressed (ELANE) regulates the LPS-induced response during injury and infection [42]. A recent study showed that ELANE participated in the maturation of neutrophil granulocytes [43]. These findings suggest that the ELANE gene may play a key role in the sepsis development.
Salmonella pathogenicity island 1 (SPI1/PU.1) is a pivotal regulator of myeloid lineage specification during hematopoiesis in mammals. Zhang and colleagues confirmed that SPI1 activation was the main cause of bone marrow suppression during sepsis [44]. Moreover, Karpurapu et al. have shown that the attenuated lung inflammation and myeloperoxidase activity were associated with favorable survival in LPS-challenged PU.1-deficient mice [45]. These results imply that suppressive SPI1 may provide a new complementary treatment option against sepsis.
C3a/C3aR axis is considered as an important pathway mediating inflammatory responses [46]. C3aR-deficient mice exhibit an increased lethality to endotoxin shock, which implied that C3aR act as a protective anti-inflammatory role in endotoxin-shock [47]. Napier and colleagues have reported that C3aR expression is dramatically enhanced in patients with severe sepsis [48]. Therefore, C3aR may contribute to endotoxemia severity and disease outcome, and may be used as an alternative therapeutic target for sepsis.
MiRNAs are involved in the regulating gene expression by degrading their target genes and weaken their translations. After reviewing previous studies, we found that numerous research studies in the field of sepsis have investigated the interconnection between miRNAs and hub genes. For instance, Li and associates indicated that downregulation of miR-204/miR-211 could result in candidemia-induced kidney injuries [49]. MiR-590 contributed to sepsis-induced acute kidney injury (AKI) expression to abrogate translation of the target genes [50]. MiR-21 has been shown to increase more than 30-fold in sepsis [51]. Besides, Zhang et al. recently reported that inhibition of miR-23b could be beneficial for polymicrobial sepsis-induced cardiac dysfunction [52]. Whereas, miR-146b protected against sepsis-induced mice myocardial injury [53]. Our present results indicated that several miRNAs, including miR-204, miR-211, miR-590, miR-21, miR-23b, and miR-146b, may play a pivotal role in pediatric sepsis. However, the functions of miR-204, miR-211, miR-590, miR-21, miR-23b, and miR-146b in pediatric sepsis need to be further defined. Additionally, research that focuses on the genes and miRNAs in pediatric sepsis are still limited.
It is evident that gene-miRNA regulatory networks greatly contribute to the pathophysiological process of pediatric sepsis, a finding that can help us better understand the mechanisms pediatric sepsis and provide effective and novel therapeutic strategies for pediatric sepsis. Nonetheless, our study was not without limitations, such as 1) only the top 9 hub genes was involved in our current study; 2) lack of research on detailed molecular mechanisms that the hub genes and miRNAs regulate in pediatric sepsis; 3) lack of research of some function studies about hub genes and miRNAs in the constructed networks. Even so, the results of this study remain meaningful, as these hub genes and miRNAs may provide more potential molecular biomarkers, and may improve early diagnosis, therapy, and prognosis of pediatric sepsis patients in the future.
Conclusions
Our findings suggested that compared with the healthy control group, the expressions of ITGAM, TLR8, IL1β, MMP9, MPO, FPR2, ELANE, SPI1, and C3AR1 in patients with sepsis were significantly upregulated, which may have an important influence on the pathophysiological mechanism of sepsis. Some potential target miRNAs (has-miR-204-5p, has-miR-211-5p, has-miR-590-5p, and has-miR-21-5p) were also predicted. Identification of these genes and miRNAs may contribute to the development of early diagnostic strategies, prognostic markers, and therapeutic targets for sepsis. However, experimental research is still necessary to validate the functions of these molecules in sepsis.
Figures
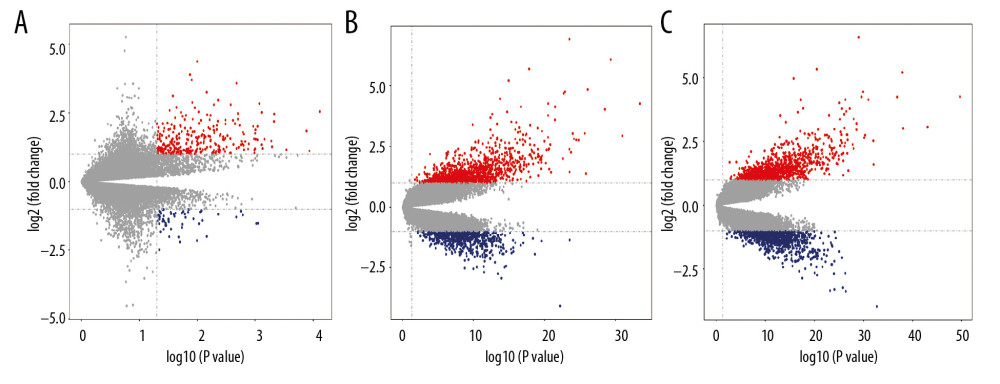 Figure 1. Volcano plot representing differential expression genes (DEGs) between control groups and sepsis groups. (A–C) Shows DEGs in GSE25504, GSE26378, and GSE26440 dataset, respectively.
Figure 1. Volcano plot representing differential expression genes (DEGs) between control groups and sepsis groups. (A–C) Shows DEGs in GSE25504, GSE26378, and GSE26440 dataset, respectively. 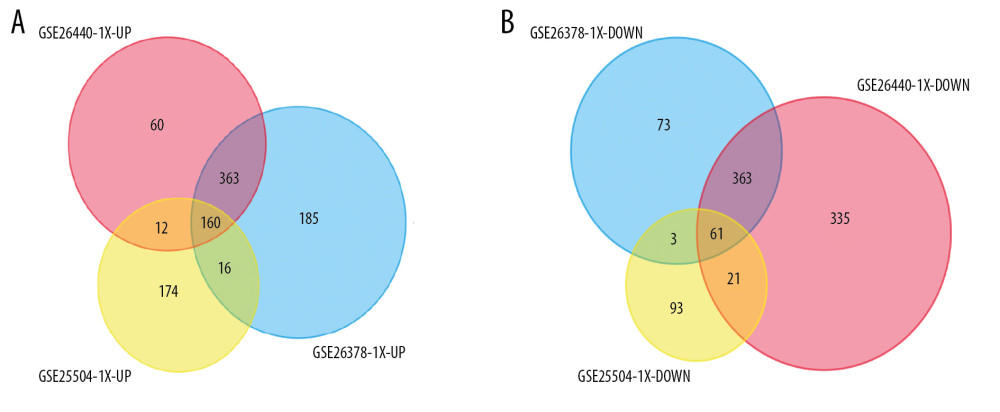 Figure 2. Venn diagrams showing the overlaps of numbers of differential expression genes (DEGs) between 3 selected Gene Expression Omnibus (GEO) datasets. (A, B) illustrate overlap of upregulated and downregulated genes in GSE25504, GSE26378, and GSE26440 dataset, respectively.
Figure 2. Venn diagrams showing the overlaps of numbers of differential expression genes (DEGs) between 3 selected Gene Expression Omnibus (GEO) datasets. (A, B) illustrate overlap of upregulated and downregulated genes in GSE25504, GSE26378, and GSE26440 dataset, respectively. 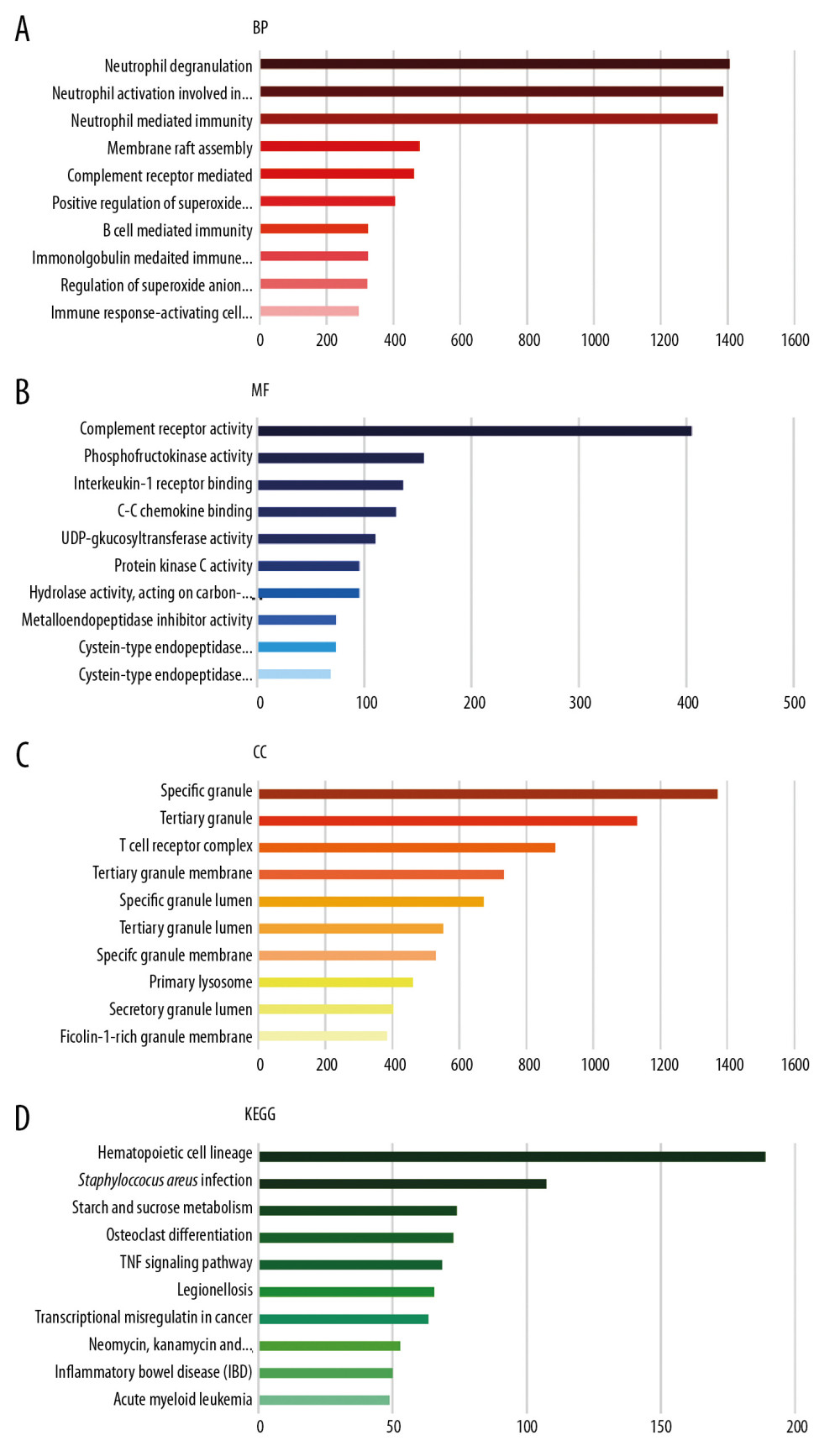 Figure 3. Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differential expression genes (DEGs). GO functional analysis showing enrichment of DEGs in (A) biological process, (B) molecular function, and (C) cellular component. (D) KEGG pathway enrichment analysis of DEGs.
Figure 3. Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differential expression genes (DEGs). GO functional analysis showing enrichment of DEGs in (A) biological process, (B) molecular function, and (C) cellular component. (D) KEGG pathway enrichment analysis of DEGs.  Figure 4. Protein–protein interaction network of 160 upregulated and 61 downregulated genes were analyzed using Cytoscape software. The network includes 219 nodes and 1062 edges. The edges between 2 nodes represent the gene-gene interactions. The size and color of the nodes corresponding to each gene were determined according to the degree of interaction. Color gradients represent the variation of the degrees of each gene from high (red) to low (blue). The closer to the red node, the higher connectivity between 2 nodes.
Figure 4. Protein–protein interaction network of 160 upregulated and 61 downregulated genes were analyzed using Cytoscape software. The network includes 219 nodes and 1062 edges. The edges between 2 nodes represent the gene-gene interactions. The size and color of the nodes corresponding to each gene were determined according to the degree of interaction. Color gradients represent the variation of the degrees of each gene from high (red) to low (blue). The closer to the red node, the higher connectivity between 2 nodes. 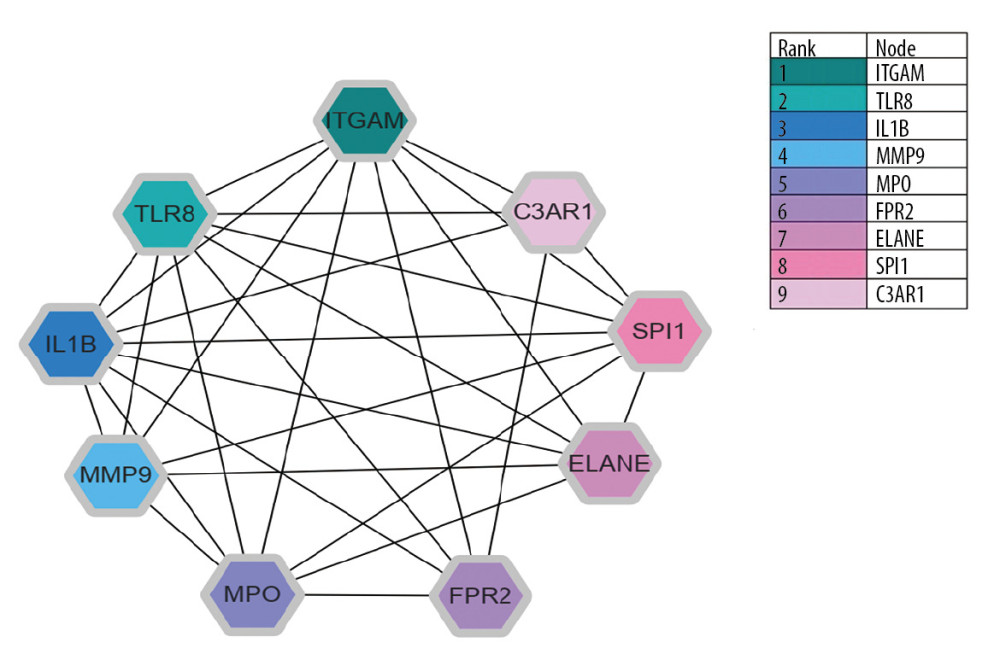 Figure 5. Protein–protein interaction network for the top 9 hub genes. Node color indicates the number of degrees. The top 9 ranked hub genes are depicted using a pseudocolor scale. Green color stands for highest degree, and pink color represents lowest degree.
Figure 5. Protein–protein interaction network for the top 9 hub genes. Node color indicates the number of degrees. The top 9 ranked hub genes are depicted using a pseudocolor scale. Green color stands for highest degree, and pink color represents lowest degree. 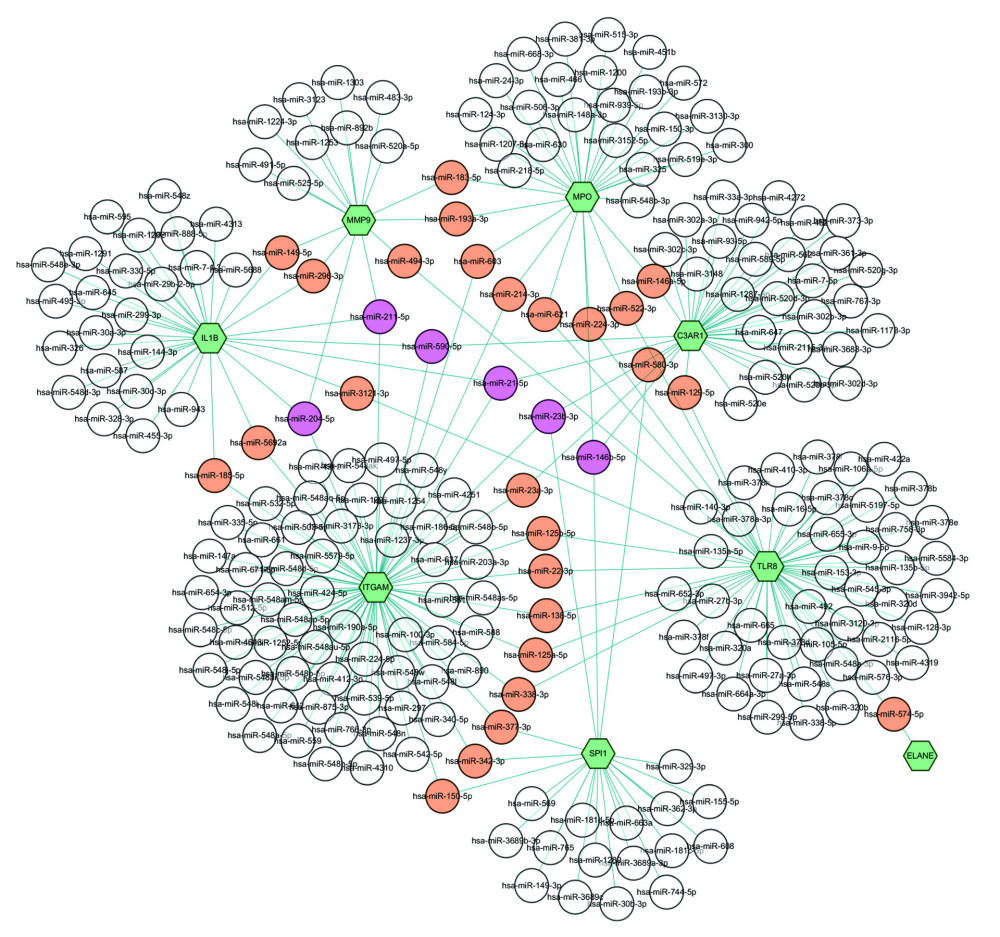 Figure 6. Integrated miRNA-DEGs networks for the top 9 hub genes. Green hexagons represent 9 hub genes. White circles represent miRNA which has low connectivity with hub genes. Red circles represent miRNA which has moderate connectivity with hub genes, and purple circles represent miRNA which has high connectivity with hub genes. miRNA-DEG – microRNA-differential expression genes.
Figure 6. Integrated miRNA-DEGs networks for the top 9 hub genes. Green hexagons represent 9 hub genes. White circles represent miRNA which has low connectivity with hub genes. Red circles represent miRNA which has moderate connectivity with hub genes, and purple circles represent miRNA which has high connectivity with hub genes. miRNA-DEG – microRNA-differential expression genes. Tables
Table 1. Details for GEO pediatric sepsis data.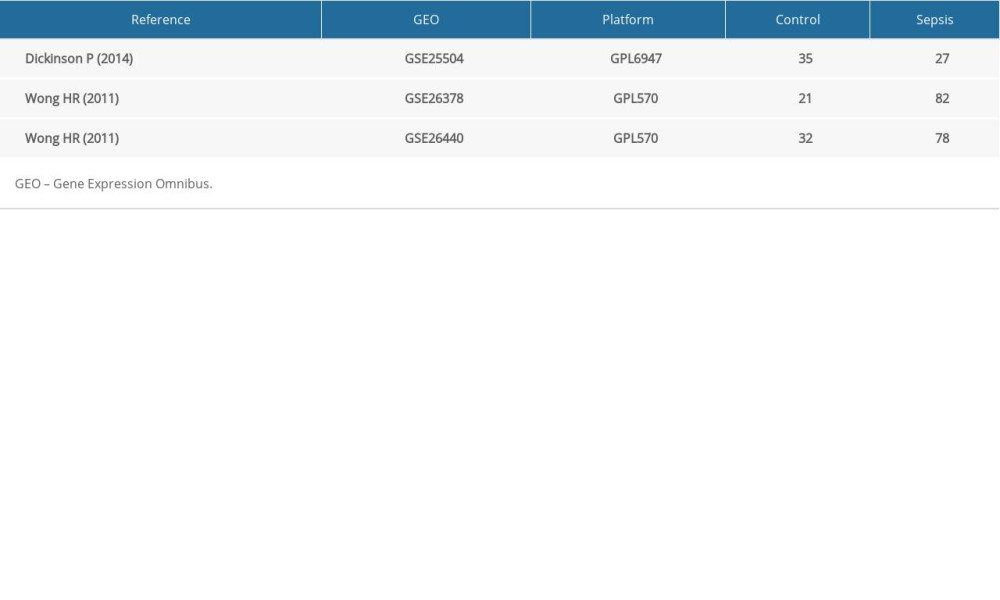 Table 2. Screening DEGs in pediatric sepsis patients by integrated microarray.
Table 2. Screening DEGs in pediatric sepsis patients by integrated microarray.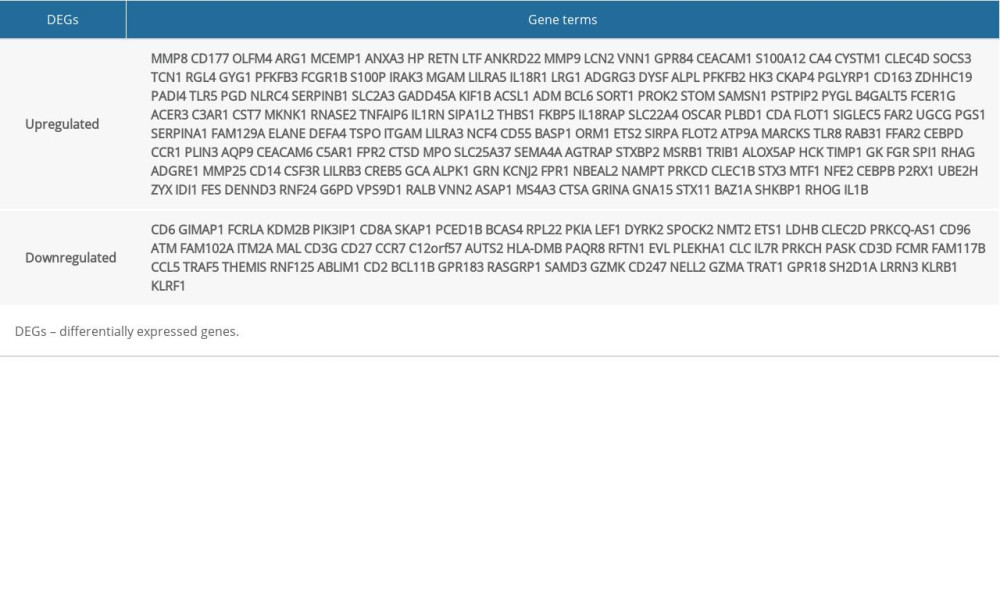 Supplementary Table 1. GO biological process terms for DEGs between the control and pediatric sepsis groups.
Supplementary Table 1. GO biological process terms for DEGs between the control and pediatric sepsis groups.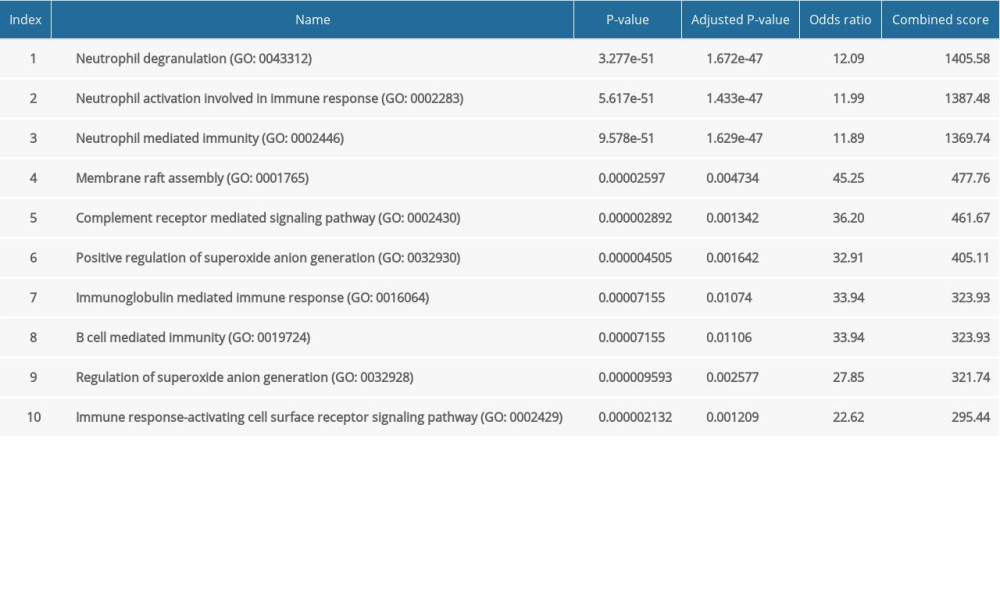 Supplementary Table 2. GO molecular function terms for DEGs between the control and pediatric sepsis groups.
Supplementary Table 2. GO molecular function terms for DEGs between the control and pediatric sepsis groups.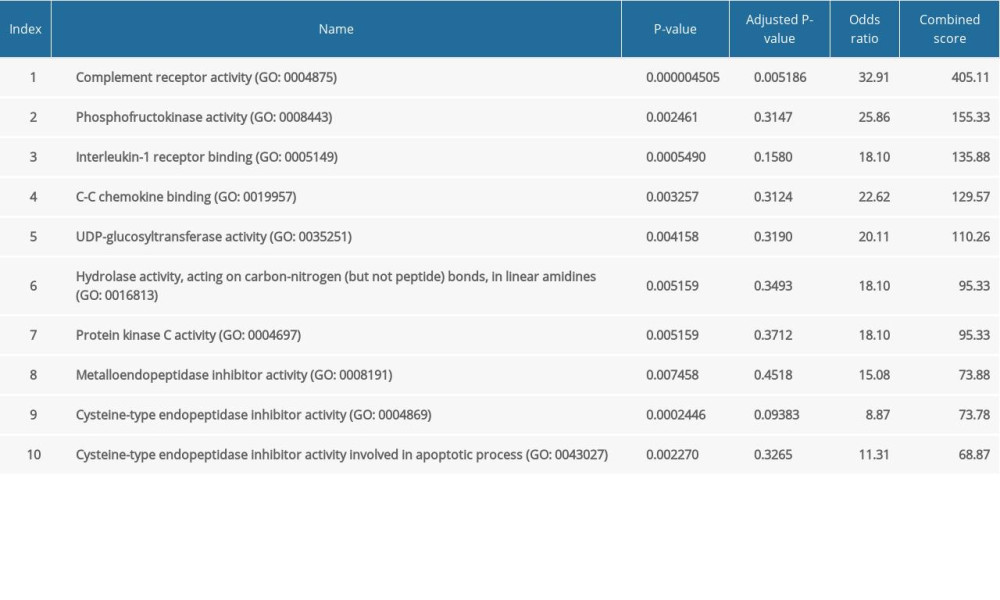 Supplementary Table 3. GO cellular component terms for DEGs between the control and pediatric sepsis groups.
Supplementary Table 3. GO cellular component terms for DEGs between the control and pediatric sepsis groups.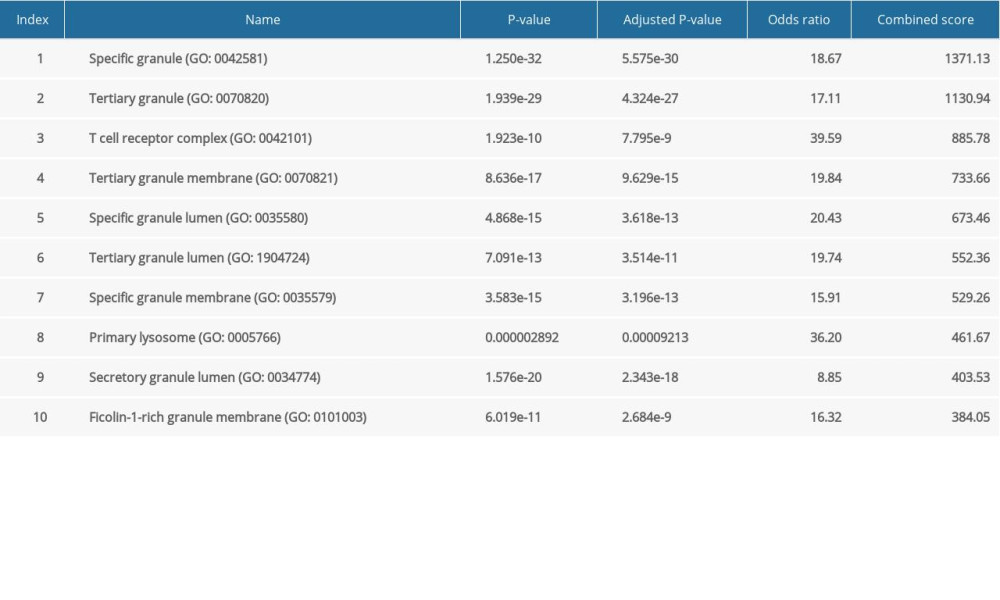 Supplementary Table 4. KEGG pathway enrichment terms for DEGs between the control and pediatric sepsis groups.
Supplementary Table 4. KEGG pathway enrichment terms for DEGs between the control and pediatric sepsis groups.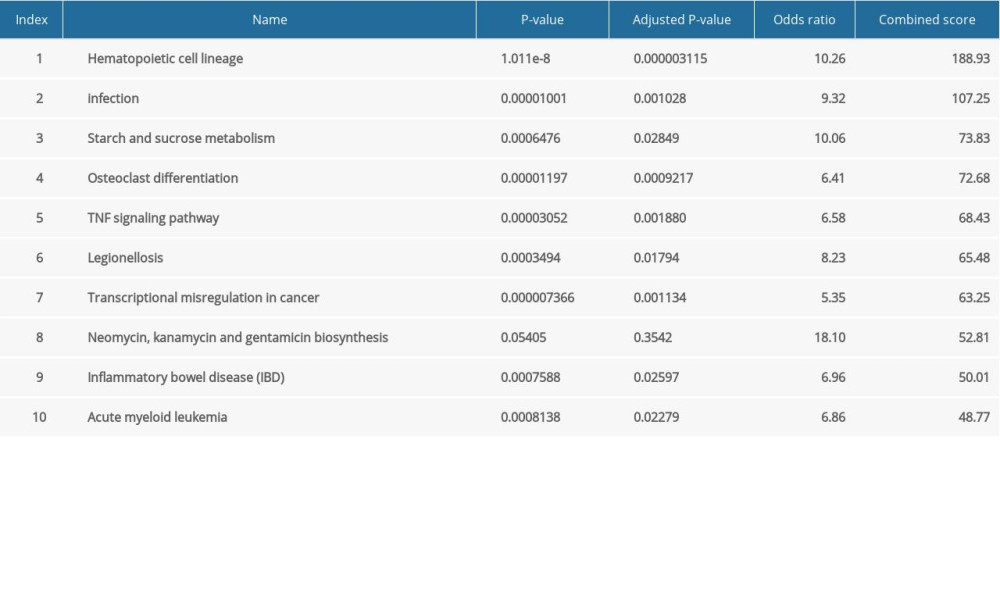
References
1. He C, Liu L, Chu Y, National and subnational all-cause and cause-specific child mortality in China, 1996–2015: A systematic analysis with implications for the Sustainable Development Goals: Lancet Glob Health, 2017; 5(2); e186-97
2. Ames SG, Davis BS, Angus DC, Hospital variation in risk-adjusted pediatric sepsis mortality: Pediatr Crit Care Med, 2018; 19(5); 390-96
3. Prout AJ, Talisa VB, Carcillo JA, Children with chronic disease bear the highest burden of pediatric sepsis: J Pediatr, 2018; 199; 194-99.e1
4. Li B, Zeng Q, Personalized identification of differentially expressed pathways in pediatric sepsis: Mol Med Rep, 2017; 16(4); 5085-90
5. Weiss SL, Peters MJ, Alhazzani W, Surviving sepsis campaign international guidelines for the management of septic shock and sepsis-associated organ dysfunction in children: Intensive Care Med, 2020; 46(Suppl 1); 10-67
6. Hu Q, Gong W, Gu J, Plasma microRNA profiles as a potential biomarker in differentiating adult-onset still’s disease from sepsis: Front Immunol, 2018; 9; 3099
7. Rao L, Song Z, Yu X, Progranulin as a novel biomarker in diagnosis of early-onset neonatal sepsis: Cytokine, 2020; 128; 155000
8. Stanski NL, Stenson EK, Cvijanovich NZ, PERSEVERE biomarkers predict severe acute kidney injury and renal recovery in pediatric septic shock: Am J Respir Crit Care Med, 2020; 201(7); 848-55
9. Wang S, Xiao C, Liu C, Identification of biomarkers of sepsis-associated acute kidney injury in pediatric patients based on UPLC-QTOF/MS: Inflammation; 2019 [Epub ahead of print]
10. Yehya N, Wong HR, Adaptation of a biomarker-based sepsis mortality risk stratification tool for pediatric acute respiratory distress syndrome: Crit Care Med, 2018; 46(1); e9-16
11. Li R, Sim I, How clinical trial data sharing platforms can advance the study of biomarkers: J Law Med Ethics, 2019; 47(3); 369-73
12. Wang X, Liotta L, Clinical bioinformatics: A new emerging science: J Clin Bioinforma, 2011; 1(1); 1
13. Tsai S, Gamblin TC, Molecular characteristics of biliary tract and primary liver tumors: Surg Oncol Clin N Am, 2019; 28(4); 685-93
14. Yan P, He Y, Xie K, In silico analyses for potential key genes associated with gastric cancer: Peer J, 2018; 6; e6092
15. Ye B, Smerin D, Gao Q, High-throughput sequencing of the immune repertoire in oncology: Applications for clinical diagnosis, monitoring, and immunotherapies: Cancer Lett, 2018; 416; 42-45
16. Cakmak HA, Demir M, MicroRNA and cardiovascular diseases: Balkan Med J, 2020; 37(2); 60-71
17. Chen L, Su W, Chen H, Proteomics for biomarker identification and clinical application in kidney disease: Adv Clin Chem, 2018; 85; 91-113
18. Geng RX, Li N, Xu Y, Identification of core biomarkers associated with outcome in glioma: Evidence from bioinformatics analysis: Dis Markers, 2018; 2018 3215958
19. Avery JT, Jimenez RV, Blake JL, Mice expressing the variant rs1143679 allele of ITGAM (CD11b) show impaired DC-mediated T cell proliferation: Mamm Genome, 2019; 30(9–10); 245-59
20. Chen L, Wang YF, Liu L, Genome-wide assessment of genetic risk for systemic lupus erythematosus and disease severity: Hum Mol Genet, 2020 [Epub ahead of print
21. Fagerholm SC, Varis M, Stefanidakis M, alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation: Blood, 2006; 108(10); 3379-86
22. Zhou H, Li Y, Gui H, Antagonism of integrin CD11b affords protection against endotoxin shock and polymicrobial sepsis via attenuation of HMGB1 nucleocytoplasmic translocation and extracellular release: J Immunol, 2018; 200(5); 1771-80
23. Bergstrom B, Aune MH, Awuh JA: J Immunol, 2015; 195(3); 1100-11
24. Kruger A, Oldenburg M, Chebrolu C, Human TLR8 senses UR/URR motifs in bacterial and mitochondrial RNA: EMBO Rep, 2015; 16(12); 1656-63
25. Cervantes JL, La Vake CJ, Weinerman B, Human TLR8 is activated upon recognition of Borrelia burgdorferi RNA in the phagosome of human monocytes: J Leukoc Biol, 2013; 94(6); 1231-41
26. Ge Y, Huang M, Yao YM, Recent advances in the biology of IL-1 family cytokines and their potential roles in development of sepsis: Cytokine Growth Factor Rev, 2019; 45; 24-34
27. Fischer J, Hans D, Lamy O, “Inflammaging” and bone in the OsteoLaus cohort”: Immun Ageing, 2020; 17; 5
28. Shakoory B, Carcillo JA, Chatham WW, Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: Reanalysis of a prior phase III trial: Crit Care Med, 2016; 44(2); 275-281
29. Jin LY, Li CF, Zhu GF, Effect of siRNA against NF-kappaB on sepsis induced acute lung injury in a mouse model: Mol Med Rep, 2014; 10(2); 631-37
30. Jordakieva G, Budge-Wolfram RM, Budinsky AC, Plasma MMP-9 and TIMP-1 levels on ICU admission are associated with 30-day survival: Wien Klin Wochenschr, 2020 [Epub ahead of print]
31. Bojic S, Kotur-Stevuljevic J, Aleksic A, Matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 in sepsis after major abdominal surgery: Dis Markers, 2018; 2018 5064684
32. Larkin CM, Hante NK, Breen EP, Role of matrix metalloproteinases 2 and 9, toll-like receptor 4 and platelet-leukocyte aggregate formation in sepsis-associated thrombocytopenia: PLoS One, 2018; 13(5); e0196478
33. Yu G, Liang Y, Zheng S, Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces oxidative stress-mediated inflammation, neuronal damage, and neural stem cell injury in a murine model of stroke: J Pharmacol Exp Ther, 2018; 364(2); 311-22
34. Liu CH, Zhang WD, Wang JJ, Senegenin ameliorate acute lung injury through reduction of oxidative stress and inhibition of inflammation in cecal ligation and puncture-induced sepsis rats: Inflammation, 2016; 39(2); 900-6
35. Cherruault Y, New deterministic methods for global optimization and applications to biomedicine: Int J Biomed Comput, 1991; 27(3–4); 215-29
36. Florentino D, Della Giustina A, de Souza Goldim MP, Early life neuroimmune challenge protects the brain after sepsis in adult rats: Neurochem Int, 2020; 135; 104712
37. Della Giustina A, Goldim MP, Danielski LG, Fish oil-rich lipid emulsion modulates neuroinflammation and prevents long-term cognitive dysfunction after sepsis: Nutrition, 2020; 70; 110417
38. Kurtoglu EL, Kayhan B, Gul M, A bioactive product lipoxin A4 attenuates liver fibrosis in an experimental model by regulating immune response and modulating the expression of regeneration genes: Turk J Gastroenterol, 2019; 30(8); 745-57
39. Zhang L, Zheng YL, Hu RH, Annexin A1 mimetic peptide AC2-26 inhibits sepsis-induced cardiomyocyte apoptosis through LXA4/PI3K/AKT signaling pathway: Curr Med Sci, 2018; 38(6); 997-1004
40. Jiang WW, Gao LL, Wu MEffect of lipoxin A4 on the expression of Toll-like receptor 4 and TNF receptor-associated factor 6 in the liver of obese rats with sepsis: Zhongguo Dang Dai Er Ke Za Zhi, 2018; 20(7); 578-84 [in Chinese]
41. Gobbetti T, Coldewey SM, Chen J, Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis: Proc Natl Acad Sci USA, 2014; 111(52); 18685-90
42. Saravanan R, Holdbrook DA, Petrlova J, Structural basis for endotoxin neutralisation and anti-inflammatory activity of thrombin-derived C-terminal peptides: Nat Commun, 2018; 9(1); 2762
43. Grabowski P, Hesse S, Hollizeck S, Proteome analysis of human neutrophil granulocytes from patients with monogenic disease using data-independent acquisition: Mol Cell Proteomics, 2019; 18(4); 760-72
44. Zhang H, Rodriguez S, Wang L, Sepsis induces hematopoietic stem cell exhaustion and myelosuppression through distinct contributions of TRIF and MYD88: Stem Cell Rep, 2016; 6(6); 940-56
45. Karpurapu M, Wang X, Deng J, Functional PU.1 in macrophages has a pivotal role in NF-kappaB activation and neutrophilic lung inflammation during endotoxemia: Blood, 2011; 118(19); 5255-66
46. Sohn YK, Son S, Choi Y, Effective inhibition of C3a-mediated pro-inflammatory response by a human C3a-specific protein binder: Biotechnol Bioeng, 2020 [Epub ahead of print]
47. Huang P, Zhou Q, Lin Q, Complement C3a induces axonal hypomyelination in the periventricular white matter through activation of WNT/beta-catenin signal pathway in septic neonatal rats experimentally induced by lipopolysaccharide: Brain Pathol, 2019 [Epub ahead of print]
48. Napier BA, Brubaker SW, Sweeney TE, Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity: J Exp Med, 2016; 213(11); 2365-82
49. Li XY, Zhang K, Jiang ZY, MiR-204/miR-211 downregulation contributes to candidemia-induced kidney injuries via derepression of Hmx1 expression: Life Sci, 2014; 102(2); 139-44
50. Ma J, Li YT, Zhang SX, MiR-590-3p Attenuates acute kidney injury by inhibiting tumor necrosis factor receptor-associated factor 6 in septic mice: Inflammation, 2019; 42(2); 637-49
51. Goodwin AJ, Guo C, Cook JA, Plasma levels of microRNA are altered with the development of shock in human sepsis: An observational study: Crit Care, 2015; 19; 440
52. Zhang H, Caudle Y, Shaikh A, Inhibition of microRNA-23b prevents polymicrobial sepsis-induced cardiac dysfunction by modulating TGIF1 and PTEN: Biomed Pharmacother, 2018; 103; 869-78
53. Wang X, Yu Y, MiR-146b protect against sepsis induced mice myocardial injury through inhibition of Notch1: J Mol Histol, 2018; 49(4); 411-17
Figures
Figure 1. Volcano plot representing differential expression genes (DEGs) between control groups and sepsis groups. (A–C) Shows DEGs in GSE25504, GSE26378, and GSE26440 dataset, respectively.Figure 2. Venn diagrams showing the overlaps of numbers of differential expression genes (DEGs) between 3 selected Gene Expression Omnibus (GEO) datasets. (A, B) illustrate overlap of upregulated and downregulated genes in GSE25504, GSE26378, and GSE26440 dataset, respectively.Figure 3. Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differential expression genes (DEGs). GO functional analysis showing enrichment of DEGs in (A) biological process, (B) molecular function, and (C) cellular component. (D) KEGG pathway enrichment analysis of DEGs.Figure 4. Protein–protein interaction network of 160 upregulated and 61 downregulated genes were analyzed using Cytoscape software. The network includes 219 nodes and 1062 edges. The edges between 2 nodes represent the gene-gene interactions. The size and color of the nodes corresponding to each gene were determined according to the degree of interaction. Color gradients represent the variation of the degrees of each gene from high (red) to low (blue). The closer to the red node, the higher connectivity between 2 nodes.Figure 5. Protein–protein interaction network for the top 9 hub genes. Node color indicates the number of degrees. The top 9 ranked hub genes are depicted using a pseudocolor scale. Green color stands for highest degree, and pink color represents lowest degree.Figure 6. Integrated miRNA-DEGs networks for the top 9 hub genes. Green hexagons represent 9 hub genes. White circles represent miRNA which has low connectivity with hub genes. Red circles represent miRNA which has moderate connectivity with hub genes, and purple circles represent miRNA which has high connectivity with hub genes. miRNA-DEG – microRNA-differential expression genes. Tables
Table 1. Details for GEO pediatric sepsis data.Table 2. Screening DEGs in pediatric sepsis patients by integrated microarray.Table 1. Details for GEO pediatric sepsis data.Table 2. Screening DEGs in pediatric sepsis patients by integrated microarray.Supplementary Table 1. GO biological process terms for DEGs between the control and pediatric sepsis groups.Supplementary Table 2. GO molecular function terms for DEGs between the control and pediatric sepsis groups.Supplementary Table 3. GO cellular component terms for DEGs between the control and pediatric sepsis groups.Supplementary Table 4. KEGG pathway enrichment terms for DEGs between the control and pediatric sepsis groups. In Press
08 Mar 2024 : Clinical Research
Evaluation of Foot Structure in Preschool Children Based on Body MassMed Sci Monit In Press; DOI: 10.12659/MSM.943765
15 Apr 2024 : Laboratory Research
The Role of Copper-Induced M2 Macrophage Polarization in Protecting Cartilage Matrix in OsteoarthritisMed Sci Monit In Press; DOI: 10.12659/MSM.943738
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952